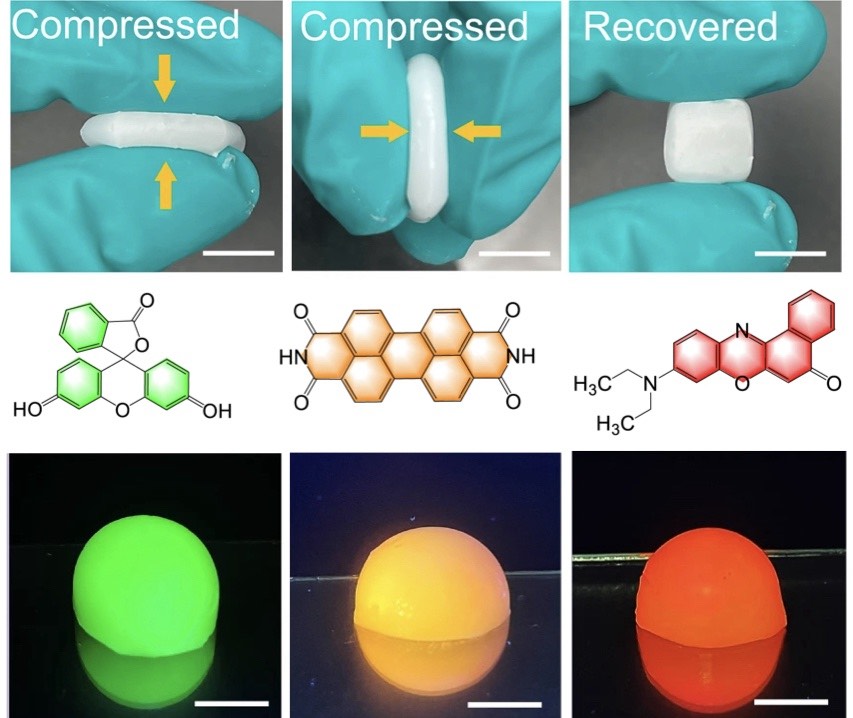
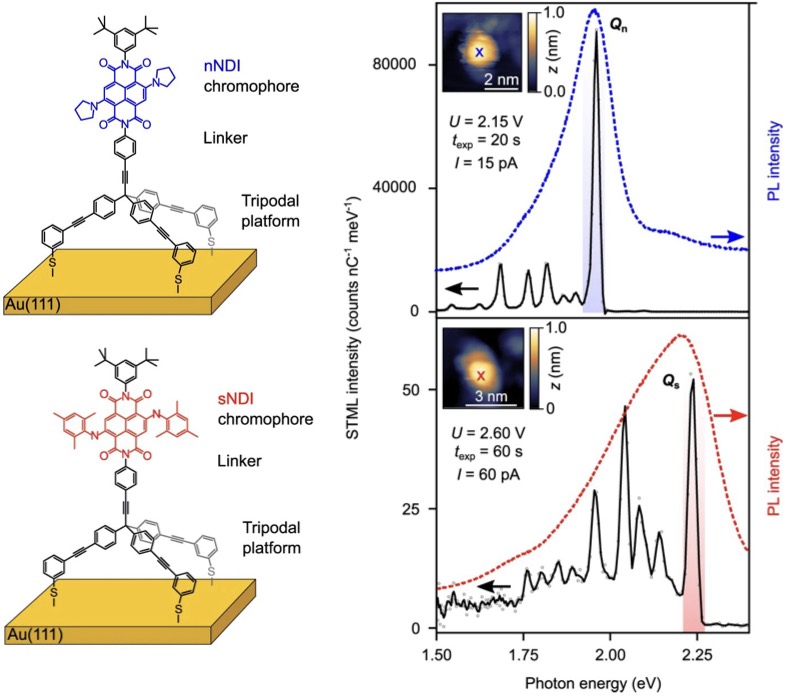
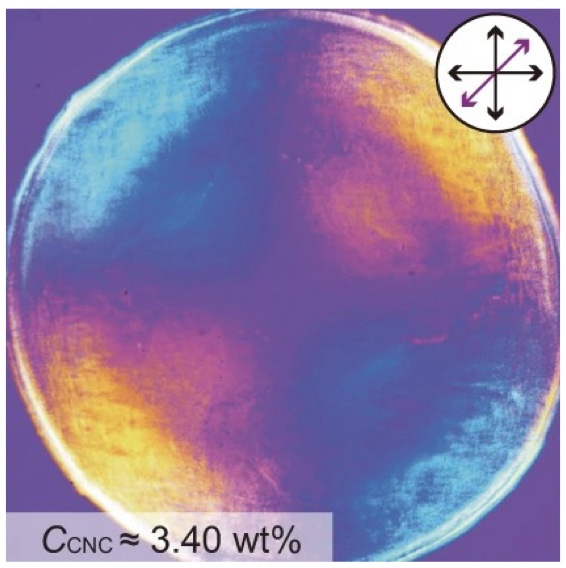
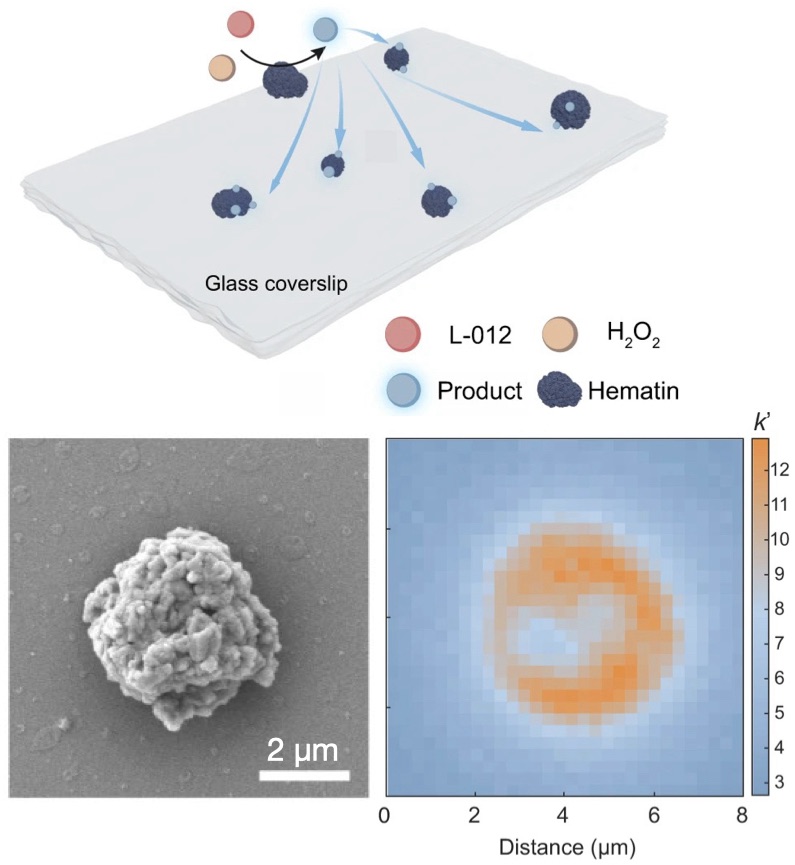
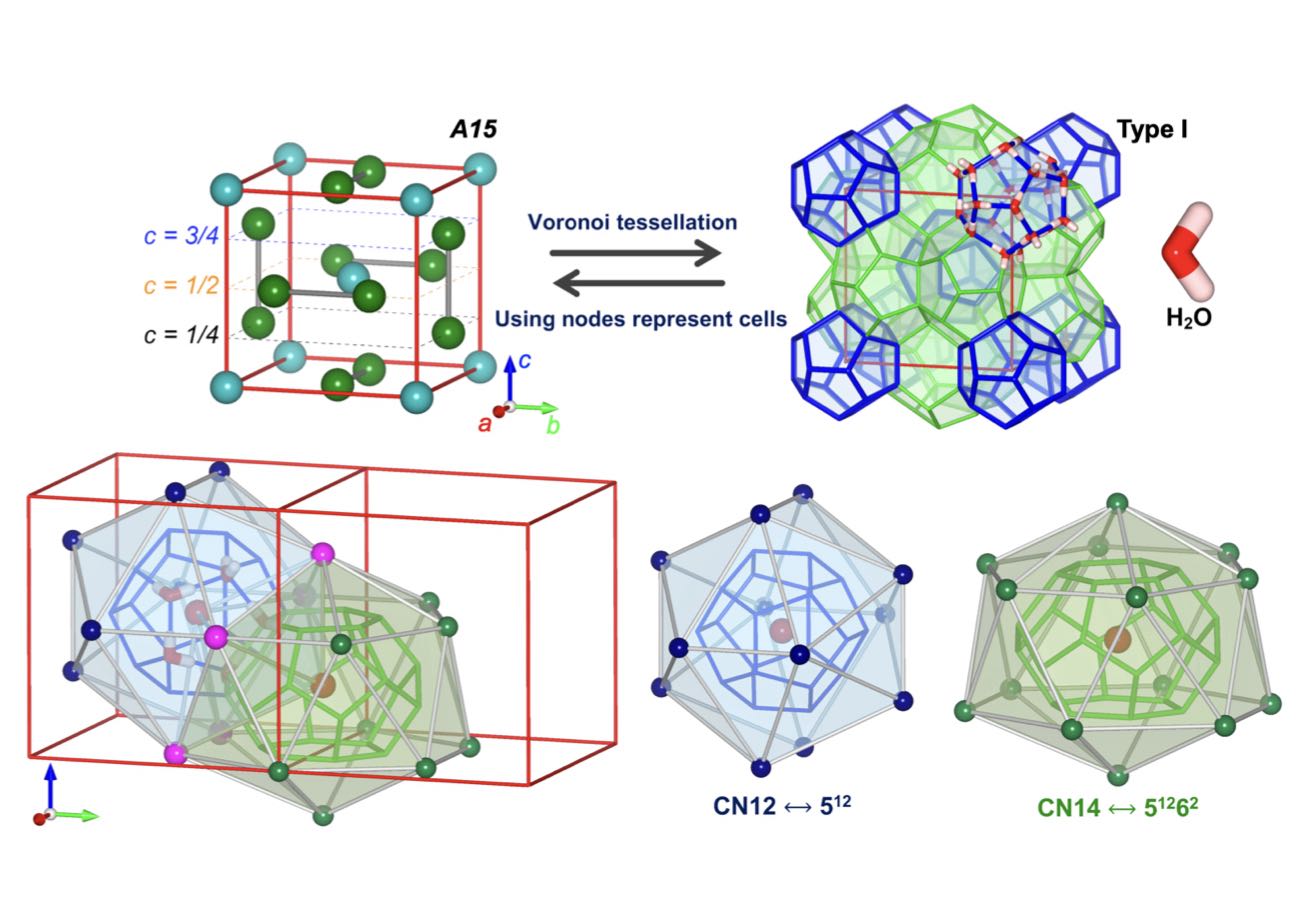
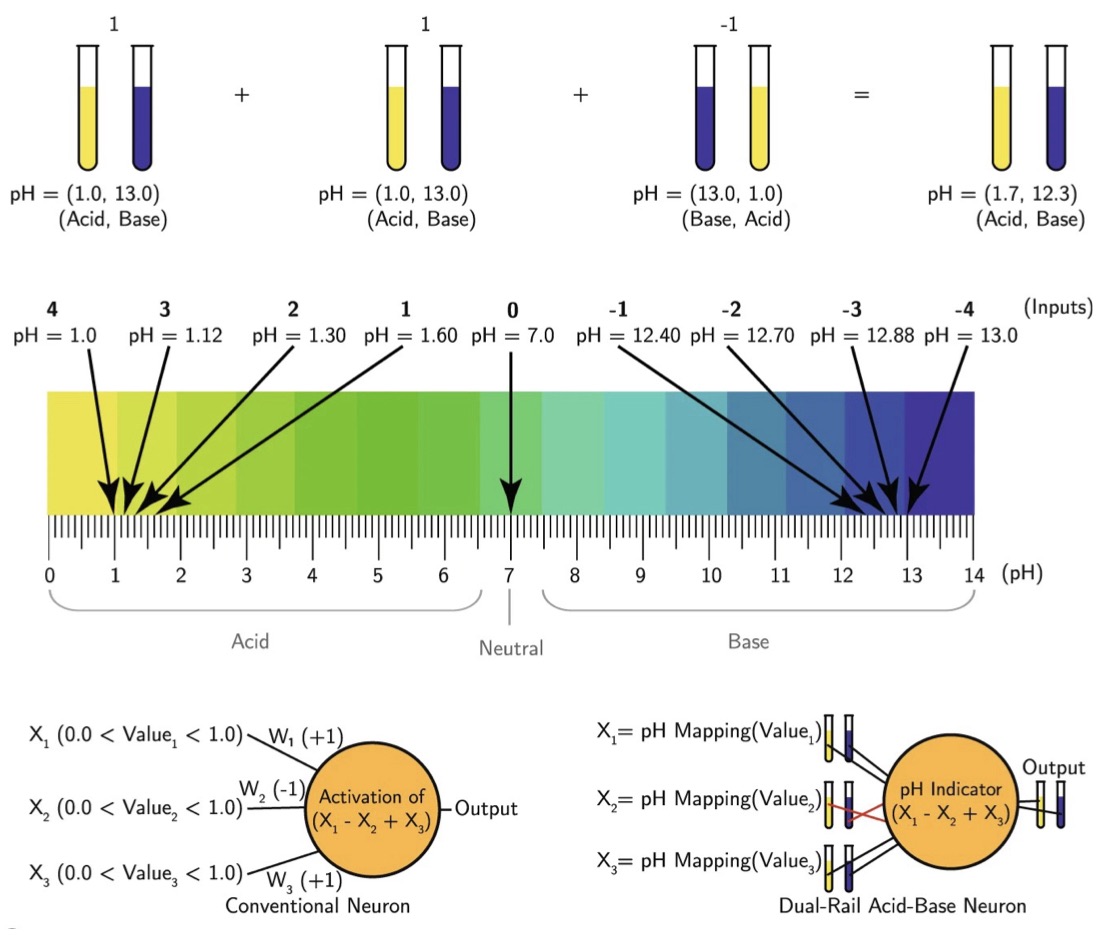
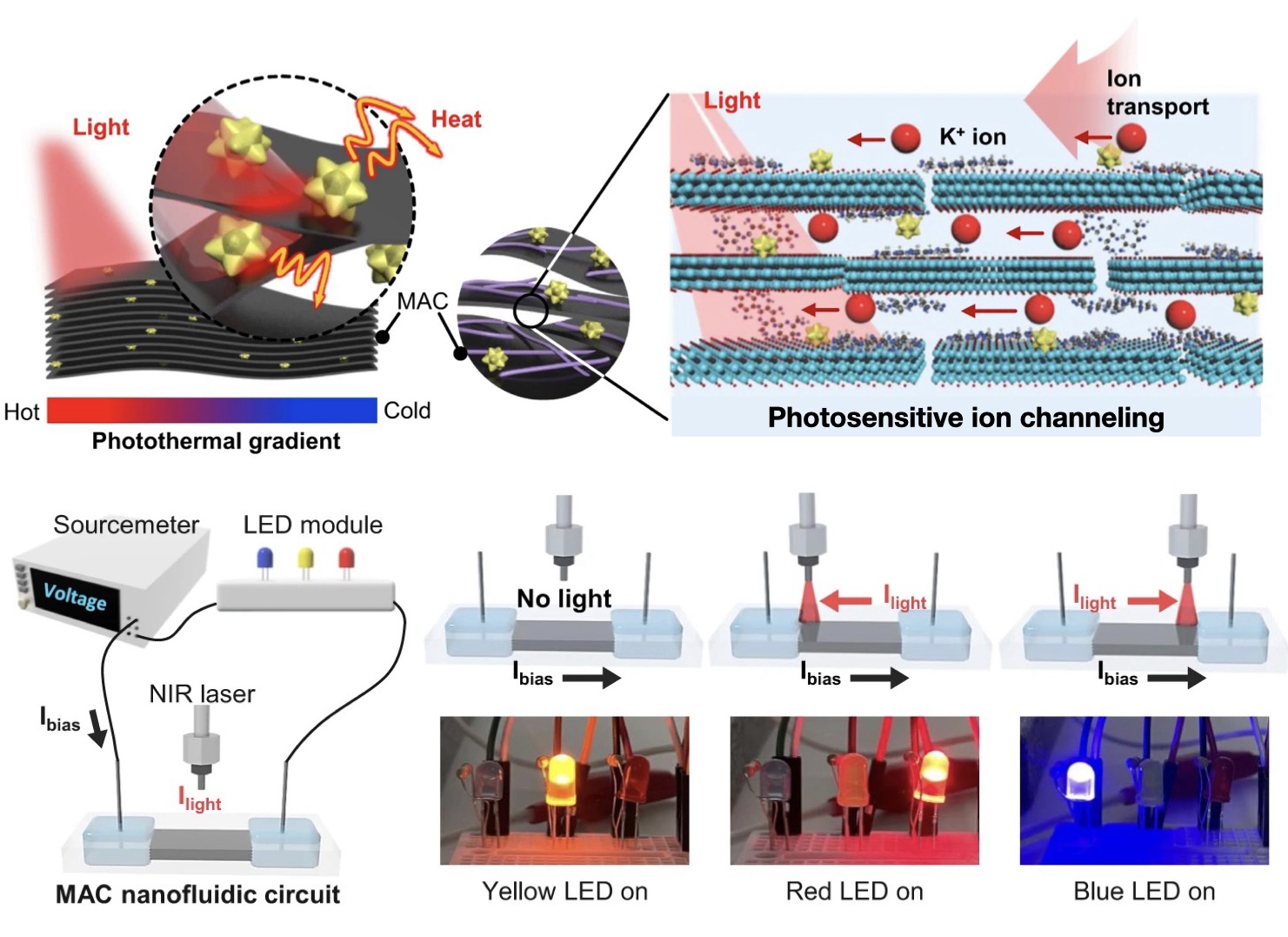
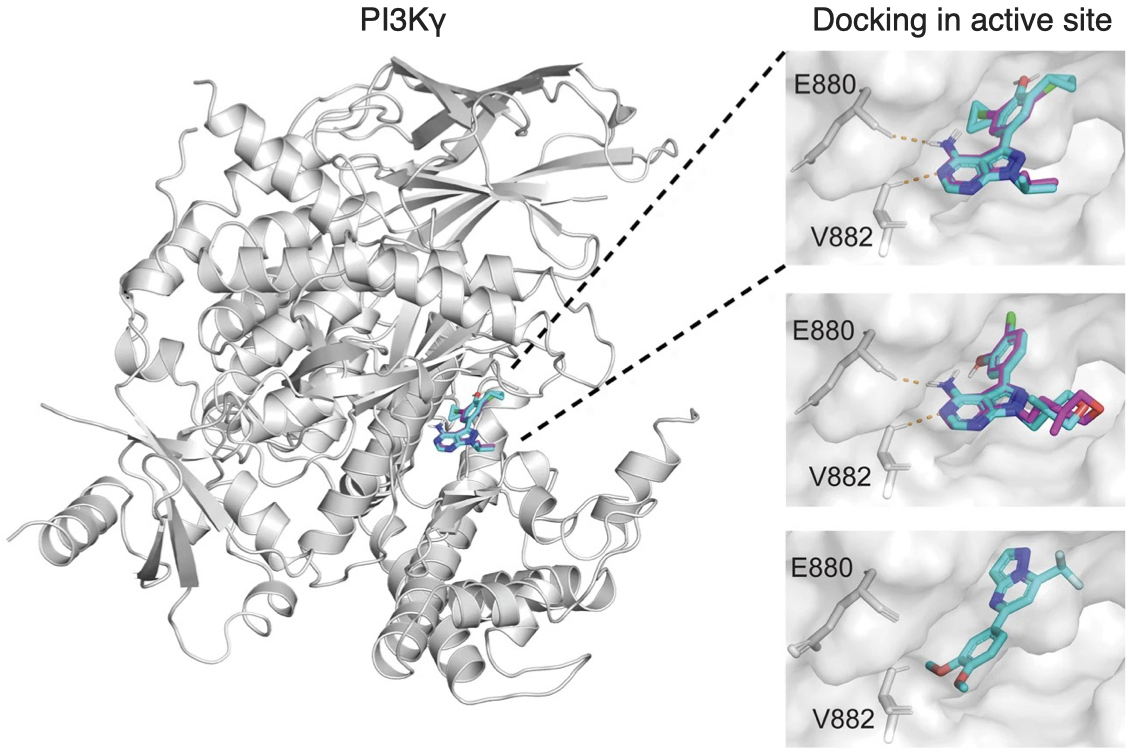
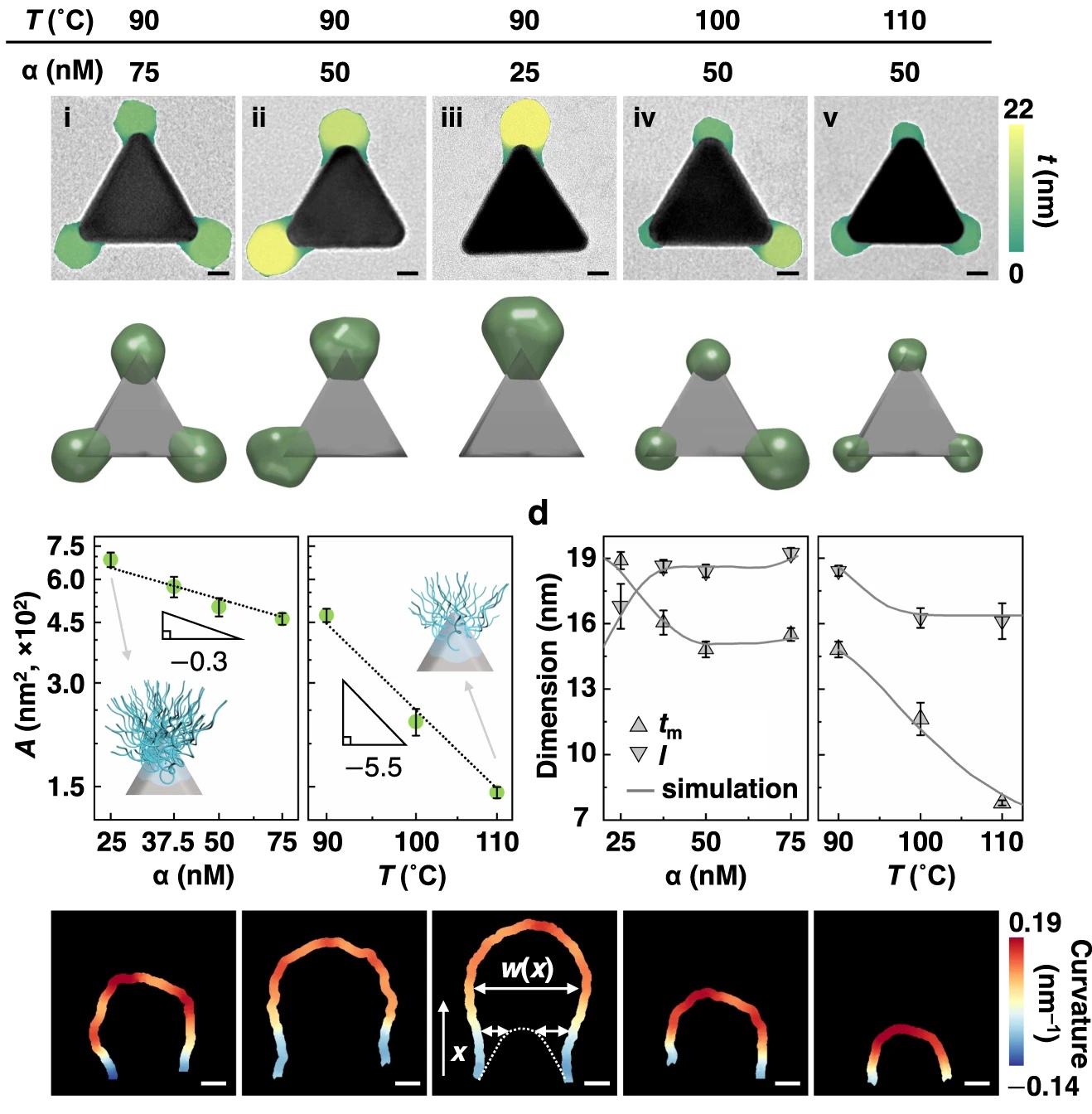
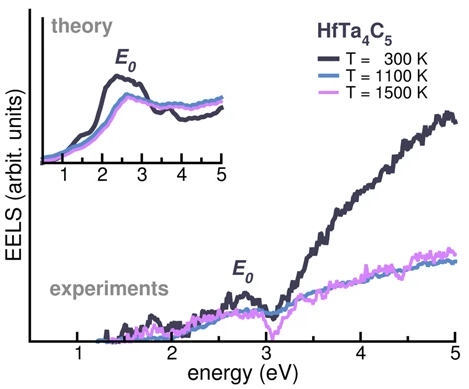
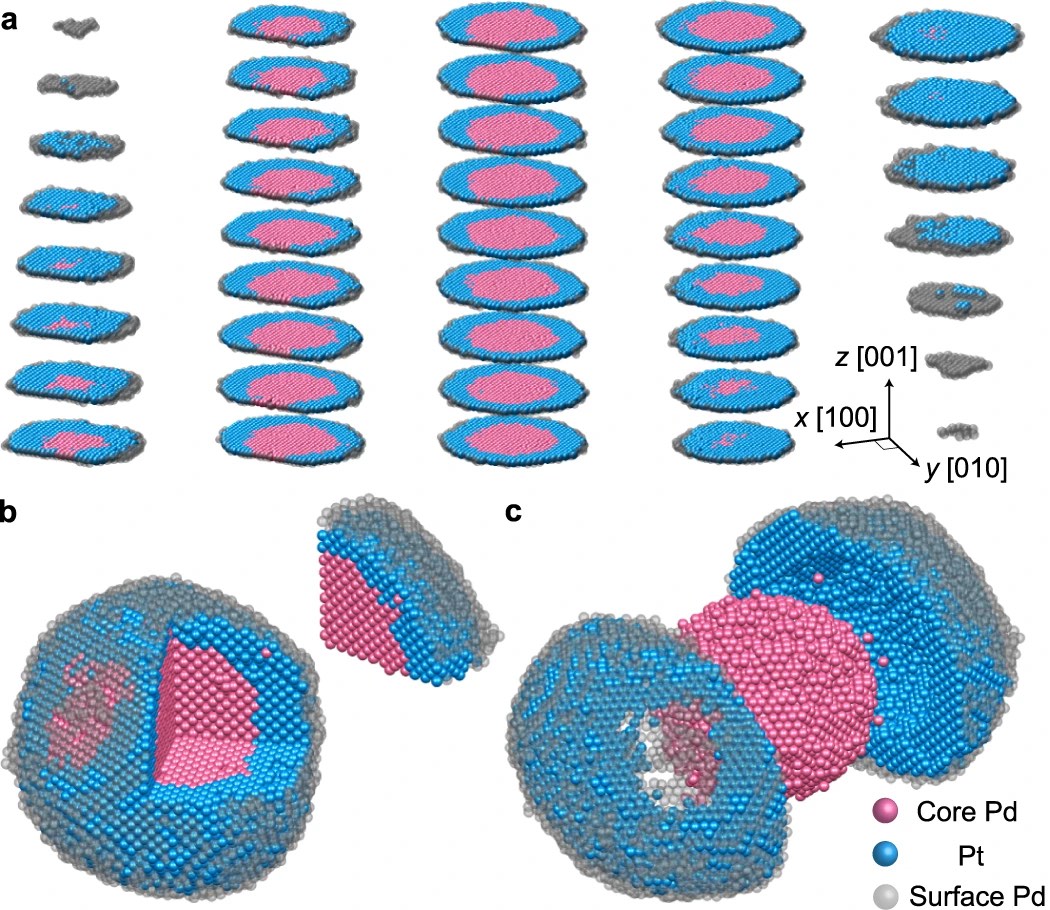
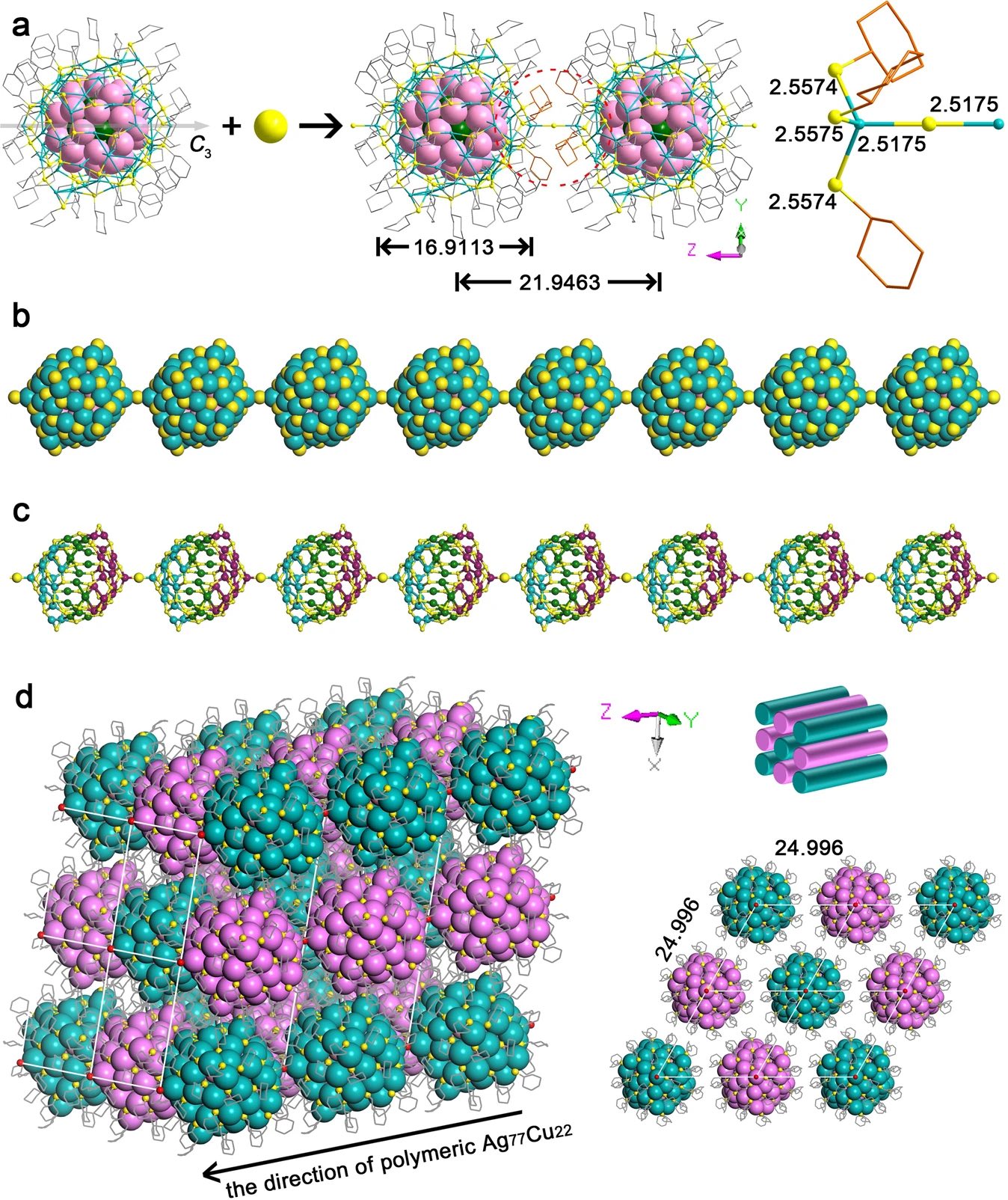
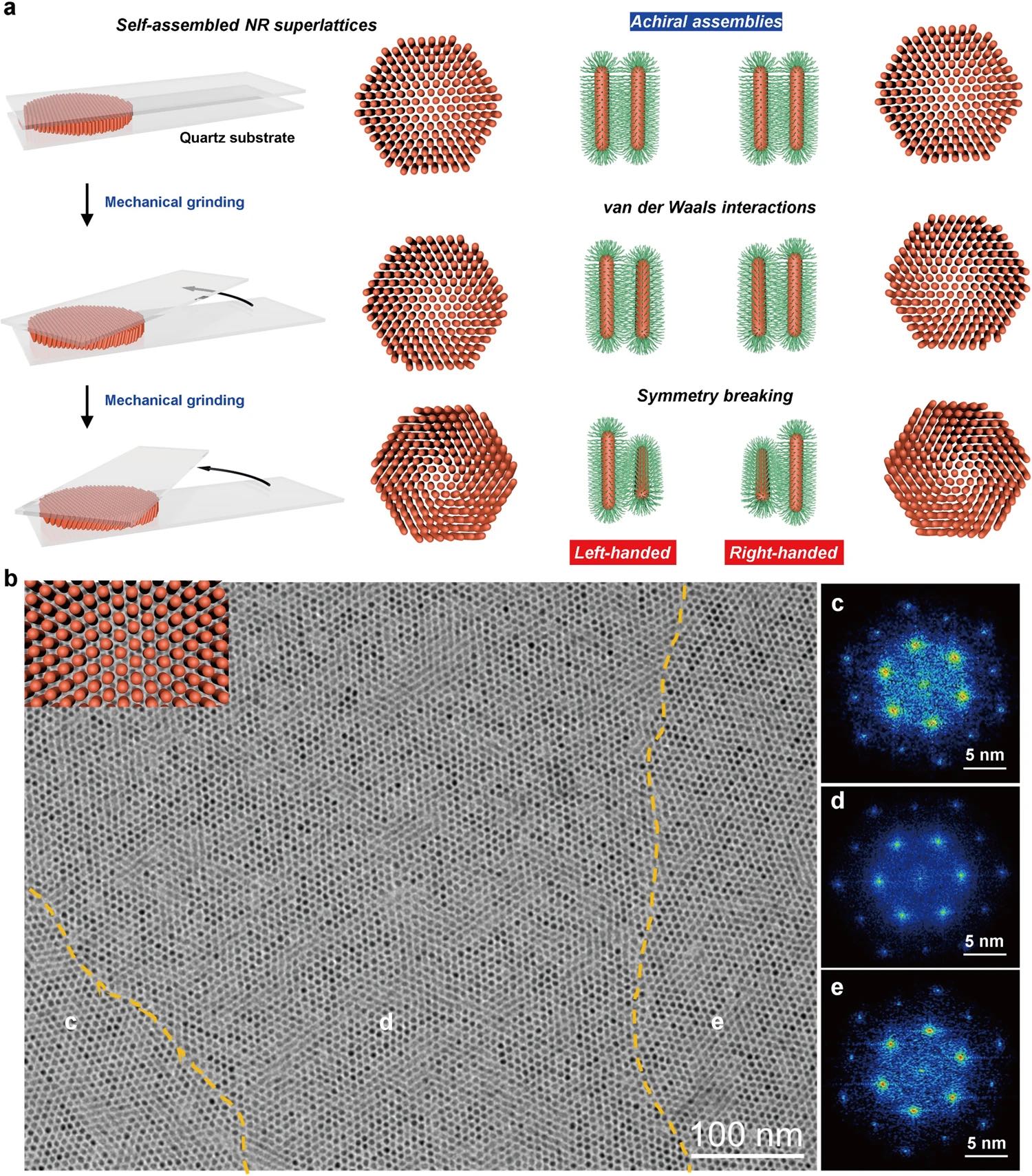
| 330. | | Xin Luo, Jun Zhu, Mirali Seyed Shariatdoust, Daniel Saliba, Felix J. Rizzuto, Reuven Gordon, R. Bruce Lennox, Hanadi F. Sleiman DNA-programmed galvanic Ostwald ripening in nanoparticle assemblies In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {DNA-programmed galvanic Ostwald ripening in nanoparticle assemblies},
author = {Xin Luo and Jun Zhu and Mirali Seyed Shariatdoust and Daniel Saliba and Felix J. Rizzuto and Reuven Gordon and R. Bruce Lennox and Hanadi F. Sleiman },
url = {https://www.nature.com/articles/s41467-026-75690-6_reference.pdf},
doi = {10.1038/s41467-026-75690-6},
year = {2026},
date = {2026-07-20},
journal = {Nature Communications},
volume = {17},
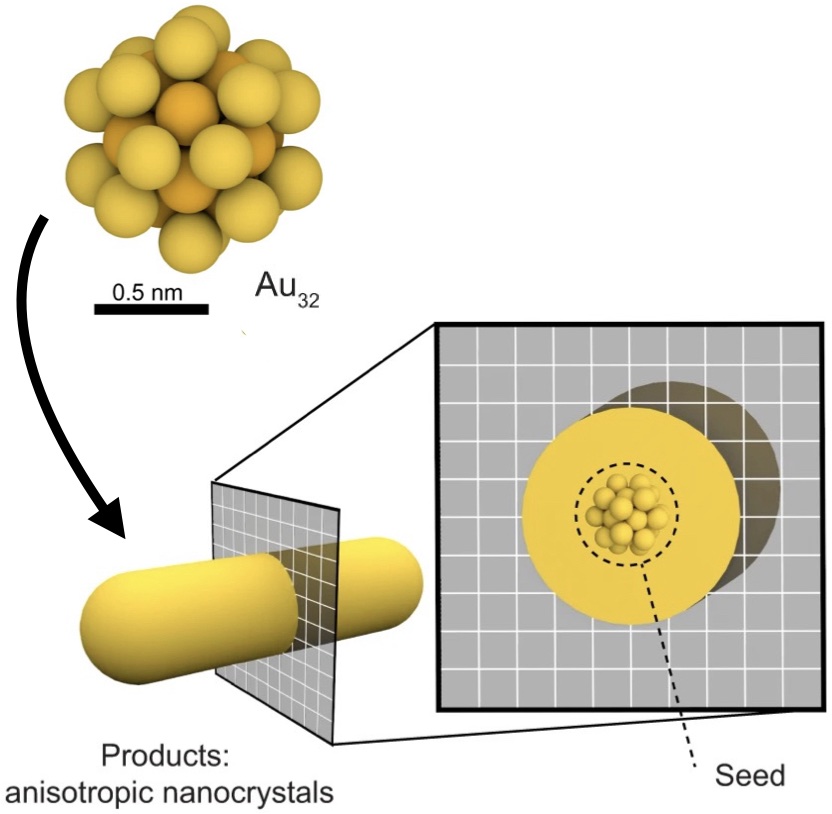
abstract = {Ostwald ripening of nanoparticles, where larger particles grow at the expense of smaller ones, is typically a thermodynamically driven process that is difficult to control. Here, we report contact-dependent, localized galvanic Ostwald ripening, a mechanism that can harness Ostwald ripening of nanoparticles as a programmable tool for nanofabrication. Using DNA origami to precisely control interparticle distances, we show that silver grows homogeneously on gold seeds until nanoparticle electrical contact triggers asymmetric ripening, driven by size-dependent electrochemical potentials. By programming when and where nanoparticles touch, we direct ripening pathways to create designer heterogeneous nanoparticle structures with enhanced plasmonic properties. In this work, we elucidate the origin of heterogeneous silver growth in nanoparticle clusters and establish design principles for engineering asymmetric nanostructures with predetermined morphologies, advancing both fundamental colloidal nanoscience and plasmonic device fabrication.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ostwald ripening of nanoparticles, where larger particles grow at the expense of smaller ones, is typically a thermodynamically driven process that is difficult to control. Here, we report contact-dependent, localized galvanic Ostwald ripening, a mechanism that can harness Ostwald ripening of nanoparticles as a programmable tool for nanofabrication. Using DNA origami to precisely control interparticle distances, we show that silver grows homogeneously on gold seeds until nanoparticle electrical contact triggers asymmetric ripening, driven by size-dependent electrochemical potentials. By programming when and where nanoparticles touch, we direct ripening pathways to create designer heterogeneous nanoparticle structures with enhanced plasmonic properties. In this work, we elucidate the origin of heterogeneous silver growth in nanoparticle clusters and establish design principles for engineering asymmetric nanostructures with predetermined morphologies, advancing both fundamental colloidal nanoscience and plasmonic device fabrication. |
| 329. | | Hao Li, Yuping Chen, Ying Xu, Yanming Liu, Qing Tang, Xi Kang, Manzhou Zhu Emergent piezoelectricity as the driving force for mechanoluminescence in crystals of atomically precise copper nanoclusters In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Emergent piezoelectricity as the driving force for mechanoluminescence in crystals of atomically precise copper nanoclusters},
author = {Hao Li and Yuping Chen and Ying Xu and Yanming Liu and Qing Tang and Xi Kang and Manzhou Zhu },
url = {https://www.nature.com/articles/s41467-026-75647-9_reference.pdf},
doi = {10.1038/s41467-026-75647-9},
year = {2026},
date = {2026-07-15},
journal = {Nature Communications},
volume = {17},
abstract = {Mechanoluminescence (ML) is a phenomenon of force-response self-luminescence, which was observed long ago. However, the underlying mechanism of ML remains ambiguous due to the complexity of light-emitting processes. Atomically precise metal nanoclusters are expected to serve as ideal model systems for unraveling ML mechanisms in specific nano-systems. Herein, we report the observation of mechanoluminescent crystals of metal nanoclusters. Cu7 clusters, which crystallize in a non-centrosymmetric trigonal space group, exhibit bright green ML when subjected to mechanical stress. Through piezoelectric experiments and theoretical calculations, we demonstrate that piezoelectricity arising from mechanical deformation of the crystal leads to an asymmetric distribution of internal dipoles within the crystal. Furthermore, crystal fracture under external force induces charge separation. Such alterations modulate the transition dipole moments and induce localized electronic transitions through strain-induced charge asymmetry, thus triggering stress-dependent luminescence. The findings provide insights into the mechanism of ML in a nanocluster system at the atomic level.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Mechanoluminescence (ML) is a phenomenon of force-response self-luminescence, which was observed long ago. However, the underlying mechanism of ML remains ambiguous due to the complexity of light-emitting processes. Atomically precise metal nanoclusters are expected to serve as ideal model systems for unraveling ML mechanisms in specific nano-systems. Herein, we report the observation of mechanoluminescent crystals of metal nanoclusters. Cu7 clusters, which crystallize in a non-centrosymmetric trigonal space group, exhibit bright green ML when subjected to mechanical stress. Through piezoelectric experiments and theoretical calculations, we demonstrate that piezoelectricity arising from mechanical deformation of the crystal leads to an asymmetric distribution of internal dipoles within the crystal. Furthermore, crystal fracture under external force induces charge separation. Such alterations modulate the transition dipole moments and induce localized electronic transitions through strain-induced charge asymmetry, thus triggering stress-dependent luminescence. The findings provide insights into the mechanism of ML in a nanocluster system at the atomic level. |
| 328. | | Bangshuo Zu, Yinfeng Long, Kaitong Deng, Lin Wang, Wenhui Wang, Mingzhu Liu, Mingjie Liu Tip functionalization of anisotropic plasmonic nanoparticles with conductive polymer patches via site-selective micelle intercalation In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Tip functionalization of anisotropic plasmonic nanoparticles with conductive polymer patches via site-selective micelle intercalation},
author = {Bangshuo Zu and Yinfeng Long and Kaitong Deng and Lin Wang and Wenhui Wang and Mingzhu Liu and Mingjie Liu },
url = {https://www.nature.com/articles/s41467-026-75238-8_reference.pdf},
doi = {10.1038/s41467-026-75238-8},
year = {2026},
date = {2026-07-13},
journal = {Nature Communications},
volume = {17},
abstract = {Plasmonic patchy nanoparticles (NPs), which consist of polymer domains distributed on metal NP surface, are important for photonics, electronics, and biomedicine applications. However, current synthetic strategies are largely limited to thiol-containing ligands or block copolymers. It remains a challenge to achieve the site-selective positioning of functional conductive polymer patches on a metal NP. Here, we show how micelle intercalation dictates the nucleation sites of polypyrrole (PPy) on anisotropic plasmonic NPs, including Au nanorods (NRs) and triangular nanoprisms. The site-selectivity of the PPy patches is governed by NP local curvature and sodium dodecyl sulphate (SDS) concentration. Moreover, we control over the charge transfer at the PPy and Au interface through HCl doping, offering possible stimuli-responsive properties at single-NP level. Furthermore, we extend the structural complexity via seed-mediated overgrowth and self-assembly. These results establish micelle intercalation as a strategy for programming conductive polymer patches on plasmonic NPs with diverse geometries.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Plasmonic patchy nanoparticles (NPs), which consist of polymer domains distributed on metal NP surface, are important for photonics, electronics, and biomedicine applications. However, current synthetic strategies are largely limited to thiol-containing ligands or block copolymers. It remains a challenge to achieve the site-selective positioning of functional conductive polymer patches on a metal NP. Here, we show how micelle intercalation dictates the nucleation sites of polypyrrole (PPy) on anisotropic plasmonic NPs, including Au nanorods (NRs) and triangular nanoprisms. The site-selectivity of the PPy patches is governed by NP local curvature and sodium dodecyl sulphate (SDS) concentration. Moreover, we control over the charge transfer at the PPy and Au interface through HCl doping, offering possible stimuli-responsive properties at single-NP level. Furthermore, we extend the structural complexity via seed-mediated overgrowth and self-assembly. These results establish micelle intercalation as a strategy for programming conductive polymer patches on plasmonic NPs with diverse geometries. |
| 327. | | Shinnosuke Hattori, Kohei Shimamura, Ken-ichi Nomura, Aiichiro Nakano, Rajiv K. Kalia, Priya Vashishta Chemical intuition on bond-dissociation energies as an emergent ability of universal machine-learning interatomic potentials In: Nature Communications, 2026, (Article in Press). @article{nokey,
title = {Chemical intuition on bond-dissociation energies as an emergent ability of universal machine-learning interatomic potentials},
author = {Shinnosuke Hattori and Kohei Shimamura and Ken-ichi Nomura and Aiichiro Nakano and Rajiv K. Kalia and Priya Vashishta },
url = {https://www.nature.com/articles/s41467-026-74919-8_reference.pdf},
doi = {10.1038/s41467-026-74919-8},
year = {2026},
date = {2026-07-02},
journal = {Nature Communications},
abstract = {Machine-learning interatomic potentials have demonstrated power-law scaling in predictive accuracy as training data and model capacity increase, but it remains unclear whether models trained at scale acquire interpretable chemical concepts. Here we show that an E(3)-equivariant machine-learning interatomic potential learns local bond information without direct supervision of bond properties. To expose this information, we develop an edge-wise emergent energy-decomposition framework and apply it to an Allegro neural-network potential trained on SPICE2, a dataset composed of stable molecular structures. The framework analyzes edge-wise energy contributions obtained from the trained model and their distributions together with information entropy to examine how data size, data composition and model-training scenarios shape the internal representation of chemical bonds. The resulting bond-dissociation energy estimates for archetypal bond types agree quantitatively with literature values and are consistent across models trained on organic and inorganic datasets. We further examine a hybrid training set and find that combining complementary chemical data improves transition-state energy-prediction accuracy while reshaping the learned bond representations. These results indicate that scalable interatomic potentials can acquire transferable bond concepts without explicit bond-energy labels, providing a way to analyze the transition-state energy-prediction problem.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Machine-learning interatomic potentials have demonstrated power-law scaling in predictive accuracy as training data and model capacity increase, but it remains unclear whether models trained at scale acquire interpretable chemical concepts. Here we show that an E(3)-equivariant machine-learning interatomic potential learns local bond information without direct supervision of bond properties. To expose this information, we develop an edge-wise emergent energy-decomposition framework and apply it to an Allegro neural-network potential trained on SPICE2, a dataset composed of stable molecular structures. The framework analyzes edge-wise energy contributions obtained from the trained model and their distributions together with information entropy to examine how data size, data composition and model-training scenarios shape the internal representation of chemical bonds. The resulting bond-dissociation energy estimates for archetypal bond types agree quantitatively with literature values and are consistent across models trained on organic and inorganic datasets. We further examine a hybrid training set and find that combining complementary chemical data improves transition-state energy-prediction accuracy while reshaping the learned bond representations. These results indicate that scalable interatomic potentials can acquire transferable bond concepts without explicit bond-energy labels, providing a way to analyze the transition-state energy-prediction problem. |
| 326. | | Haitao Zhang, Ryo Nakanishi, Takefumi Yoshida, Masahiko Nishijima, Carmen Herrmann, Masahiro Yamashita Confined ionic order in atomic nanowires of rare-earth chlorides unveiled via symmetry-guided structural screening In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Confined ionic order in atomic nanowires of rare-earth chlorides unveiled via symmetry-guided structural screening},
author = {Haitao Zhang and Ryo Nakanishi and Takefumi Yoshida and Masahiko Nishijima and Carmen Herrmann and Masahiro Yamashita },
url = {https://www.nature.com/articles/s41467-026-74793-4_reference.pdf},
doi = {10.1038/s41467-026-74793-4},
year = {2026},
date = {2026-06-27},
urldate = {2026-06-27},
journal = {Nature Communications},
volume = {17},
abstract = {Low-dimensional materials can exhibit unusual structural, electronic, and magnetic properties that arise from nanoscale confinement and quantum effects. However, determining their local atomic structures remains challenging, especially when long-range periodicity is limited and conventional crystallographic approaches are difficult to apply. Here, we show a symmetry-guided screening strategy for resolving confined one-dimensional materials by combining scanning transmission electron microscopy with reactive force-field molecular dynamics and density functional theory. We encapsulate rare-earth chlorides within single-walled carbon nanotubes (SWCNTs) to form stable LnCl3@SWCNTs hybrids (Ln = Y, Gd, Dy, Er), which contain ordered one-dimensional atomic nanowires. The screening strategy identifies structural models that are consistent with experimental imaging and energetic stability, enabling local configurations to be distinguished. Electronic and magnetic analyses, particularly for Dy-based systems using complete active space self-consistent field calculations and Boltzmann transport theory, reveal how local symmetry and confinement influence their functional properties. This experiment–theory framework provides a broadly applicable route for probing short-range-ordered nanostructures, establishing structure–property correlations, and guiding the design of functional low-dimensional materials.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Low-dimensional materials can exhibit unusual structural, electronic, and magnetic properties that arise from nanoscale confinement and quantum effects. However, determining their local atomic structures remains challenging, especially when long-range periodicity is limited and conventional crystallographic approaches are difficult to apply. Here, we show a symmetry-guided screening strategy for resolving confined one-dimensional materials by combining scanning transmission electron microscopy with reactive force-field molecular dynamics and density functional theory. We encapsulate rare-earth chlorides within single-walled carbon nanotubes (SWCNTs) to form stable LnCl3@SWCNTs hybrids (Ln = Y, Gd, Dy, Er), which contain ordered one-dimensional atomic nanowires. The screening strategy identifies structural models that are consistent with experimental imaging and energetic stability, enabling local configurations to be distinguished. Electronic and magnetic analyses, particularly for Dy-based systems using complete active space self-consistent field calculations and Boltzmann transport theory, reveal how local symmetry and confinement influence their functional properties. This experiment–theory framework provides a broadly applicable route for probing short-range-ordered nanostructures, establishing structure–property correlations, and guiding the design of functional low-dimensional materials. |
| 325. | | Kai Wei, Kai Chang, Xinyi Wang, Shoufeng Lan, M. Cynthia Hipwell, Heng Pan 3D nanoprinting of metals by spatiotemporally confined hot electrons via multiple-electron excitations in nanocrystals In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {3D nanoprinting of metals by spatiotemporally confined hot electrons via multiple-electron excitations in nanocrystals},
author = {Kai Wei and Kai Chang and Xinyi Wang and Shoufeng Lan and M. Cynthia Hipwell and Heng Pan },
url = {https://www.nature.com/articles/s41467-026-74926-9_reference.pdf},
doi = {10.1038/s41467-026-74926-9},
year = {2026},
date = {2026-06-27},
journal = {Nature Communications},
volume = {17},
abstract = {For decades, multi-photon polymerization has been a key technology for fabricating complex 3D micro- and nanoscale structures from polymer materials. However, extending this capability beyond polymers remains a significant challenge. In particular, nanoscale metal printing is challenging because solid metal formation typically requires energy-driven reactions or thermally activated processes that are difficult to spatially localize, resulting in compromised feature resolution. Here, we demonstrate 3D nanoprinting of metals with depth and lateral resolution <250 nm. The method employs femtosecond laser-induced hot electrons spatiotemporally confined in nanocrystals to facilitate nonlinear multi-electron absorption, ligand desorption, and nanocrystal fusion. This approach operates at a pulse energy about 100× lower than simultaneous multi-photon processes, avoids organic additives, and is compatible with free-space or layer-by-layer printing. Printing of multiple metals is demonstrated, achieving mechanical strength comparable to pure metals, along with functional mechanical and optical metamaterials. This technology enables customizable 3D metal nanoprinting for advanced applications in metamaterials, biotechnology, nanorobotics, sensors, and semiconductor manufacturing.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
For decades, multi-photon polymerization has been a key technology for fabricating complex 3D micro- and nanoscale structures from polymer materials. However, extending this capability beyond polymers remains a significant challenge. In particular, nanoscale metal printing is challenging because solid metal formation typically requires energy-driven reactions or thermally activated processes that are difficult to spatially localize, resulting in compromised feature resolution. Here, we demonstrate 3D nanoprinting of metals with depth and lateral resolution <250 nm. The method employs femtosecond laser-induced hot electrons spatiotemporally confined in nanocrystals to facilitate nonlinear multi-electron absorption, ligand desorption, and nanocrystal fusion. This approach operates at a pulse energy about 100× lower than simultaneous multi-photon processes, avoids organic additives, and is compatible with free-space or layer-by-layer printing. Printing of multiple metals is demonstrated, achieving mechanical strength comparable to pure metals, along with functional mechanical and optical metamaterials. This technology enables customizable 3D metal nanoprinting for advanced applications in metamaterials, biotechnology, nanorobotics, sensors, and semiconductor manufacturing. |
| 324. | | Ville Liljeström, Pietro Castronovo, Daisy Agrawal, Susobhan Das, Negar Hosseiniyan, Jani Seitsonen, Hua Jiang, Marco Cannas, Alice Sciortino, Zhipei Sun, Fabrizio Messina, Sourov Chandra Hierarchical self-assembly of atomically precise Au6 nanoclusters into fibrillar superstructures with collective optical properties In: Nature Communications, 2026, (Article in Press). @article{nokey,
title = {Hierarchical self-assembly of atomically precise Au6 nanoclusters into fibrillar superstructures with collective optical properties},
author = {Ville Liljeström and Pietro Castronovo and Daisy Agrawal and Susobhan Das and Negar Hosseiniyan and Jani Seitsonen and Hua Jiang and Marco Cannas and Alice Sciortino and Zhipei Sun and Fabrizio Messina and Sourov Chandra },
url = {https://www.nature.com/articles/s41467-026-74923-y_reference.pdf},
doi = {10.1038/s41467-026-74923-y},
year = {2026},
date = {2026-06-27},
urldate = {2026-06-27},
journal = {Nature Communications},
abstract = {Self-assembly, a key approach in material science, enables modulation of nanostructures to achieve distinct materials properties. Atomically precise metal nanoclusters (NCs), consisting of a few metal atoms, exhibit distinctive optical and electronic properties due to their “molecule-like” discrete energy levels. Such NCs offer advantages over conventional metal nanoparticles by avoiding dispersity in size and uncontrolled aggregation. Here we demonstrate a study on crystalline aggregates of Au6 NCs, formed under varying conditions. Notably, the controlled aggregation of these Au6 NCs, synchronising by protonation or employing specific hydrogen bonding, yields self-assembled nanoribbons and percolated networks of nanofibers that further produce extended 3D fibrillar architectures. X-ray scattering and electron microscopy reveal two distinct packing modes: a monoclinic 2D oblique lattice with sparse NC arrangement, and a nematic 2D hexagonal packing, resembling liquid-crystalline rod-like assemblies. The resulting superstructures exhibit enhanced optical responses, retaining the photoluminescence of their constituents, and manifesting third-harmonic generation, slow photoluminescence decay driven by charge-migration effects, and polarization effects influenced by the ordered structure with the periodicity of the NCs. Overall, this work emphasizes the potential of NCs as versatile building blocks for tunable and responsive optoelectronic materials, providing insights into the mechanisms of self-assembly.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Self-assembly, a key approach in material science, enables modulation of nanostructures to achieve distinct materials properties. Atomically precise metal nanoclusters (NCs), consisting of a few metal atoms, exhibit distinctive optical and electronic properties due to their “molecule-like” discrete energy levels. Such NCs offer advantages over conventional metal nanoparticles by avoiding dispersity in size and uncontrolled aggregation. Here we demonstrate a study on crystalline aggregates of Au6 NCs, formed under varying conditions. Notably, the controlled aggregation of these Au6 NCs, synchronising by protonation or employing specific hydrogen bonding, yields self-assembled nanoribbons and percolated networks of nanofibers that further produce extended 3D fibrillar architectures. X-ray scattering and electron microscopy reveal two distinct packing modes: a monoclinic 2D oblique lattice with sparse NC arrangement, and a nematic 2D hexagonal packing, resembling liquid-crystalline rod-like assemblies. The resulting superstructures exhibit enhanced optical responses, retaining the photoluminescence of their constituents, and manifesting third-harmonic generation, slow photoluminescence decay driven by charge-migration effects, and polarization effects influenced by the ordered structure with the periodicity of the NCs. Overall, this work emphasizes the potential of NCs as versatile building blocks for tunable and responsive optoelectronic materials, providing insights into the mechanisms of self-assembly. |
| 323. | | Wei Li, Tianran Zhang, Zhisheng Wang, Yufeng Wang Two-dimensional surface melting with an intermediate quasi-hexatic layer In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Two-dimensional surface melting with an intermediate quasi-hexatic layer},
author = {Wei Li and Tianran Zhang and Zhisheng Wang and Yufeng Wang },
url = {https://www.nature.com/articles/s41467-026-74678-6_reference.pdf},
doi = {10.1038/s41467-026-74678-6},
year = {2026},
date = {2026-06-23},
journal = {Nature Communications},
volume = {17},
abstract = {Melting in two-dimensional (2D) systems exhibits physics absent in three dimensions, yet its mechanism has been debated for decades. The prevailing Kosterlitz-Thouless-Halperin-Nelson-Young (KTHNY) theory describes 2D melting as two continuous transitions in the bulk with an intermediate hexatic phase. While this framework can describe bulk behavior at the transition, it does not account for the influence of free surfaces. Here, using single-particle imaging of optically-driven 2D colloidal solids, we report that melting with free surfaces proceeds via a double wetting pathway, where an inhomogeneous layer with hexatic order forms between the bulk solid and an outer quasi-liquid layer. Prior to melting, the thicknesses of both wetting layers grow into the bulk following a power-law divergence. The quasi-hexatic layer penetrates the bulk ahead of the quasi-liquid layer, giving rise to separate solid-hexatic and hexatic-liquid transitions. Different from the KTHNY scenario, in which dislocations nucleate as bound pairs in the bulk, the free surface acts as a prolific source that continuously emits dislocations into the solid, driving the invasion of the wetting layers. These results present a surface-initiated pathway for 2D melting, offering insights into colloidal matter, 2D materials, and condensed matter physics.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Melting in two-dimensional (2D) systems exhibits physics absent in three dimensions, yet its mechanism has been debated for decades. The prevailing Kosterlitz-Thouless-Halperin-Nelson-Young (KTHNY) theory describes 2D melting as two continuous transitions in the bulk with an intermediate hexatic phase. While this framework can describe bulk behavior at the transition, it does not account for the influence of free surfaces. Here, using single-particle imaging of optically-driven 2D colloidal solids, we report that melting with free surfaces proceeds via a double wetting pathway, where an inhomogeneous layer with hexatic order forms between the bulk solid and an outer quasi-liquid layer. Prior to melting, the thicknesses of both wetting layers grow into the bulk following a power-law divergence. The quasi-hexatic layer penetrates the bulk ahead of the quasi-liquid layer, giving rise to separate solid-hexatic and hexatic-liquid transitions. Different from the KTHNY scenario, in which dislocations nucleate as bound pairs in the bulk, the free surface acts as a prolific source that continuously emits dislocations into the solid, driving the invasion of the wetting layers. These results present a surface-initiated pathway for 2D melting, offering insights into colloidal matter, 2D materials, and condensed matter physics. |
| 322. | | Wei Guo, Tzu-Heng Chen, Nathan Ronceray, Eveline Mayner, Kenji Watanabe, Takashi Taniguchi, Aleksandra Radenovic Emission dipole orientation reveals dynamic single-molecule interactions with 2D crystals at solvent interfaces In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Emission dipole orientation reveals dynamic single-molecule interactions with 2D crystals at solvent interfaces},
author = {Wei Guo and Tzu-Heng Chen and Nathan Ronceray and Eveline Mayner and Kenji Watanabe and Takashi Taniguchi and Aleksandra Radenovic},
url = {https://www.nature.com/articles/s41467-026-74191-w_reference.pdf},
doi = {10.1038/s41467-026-74191-w},
year = {2026},
date = {2026-06-16},
journal = {Nature Communications},
volume = {17},
abstract = {Direct observation of individual fluorescent emitters is essential for studying quantum materials, chemical reactions, and biological systems. However, current single-molecule tracking methods only focuses on the localizations of molecules, overlooking molecular configuration and orientation. In this work, we introduce a high-throughput polarized single-molecule localization microscopy that simultaneously resolves the locations and emission dipole orientations of single fluorescent emitters with nanometer precision. Using the interface between pristine hexagonal boron nitride (h-BN) and an organic solvent as a challenging platform, we capture over 10⁵ fluorescent events and reveal distinct molecular interaction dynamics at room temperature. The measured dipole orientations align with the three-fold (C₃) rotational symmetry of the h-BN lattice, and molecular dynamics in the liquid environment can be modulated electrochemically, suggesting a route for on-demand control of quantum emitters. We also find that lateral diffusion at the solid–liquid interface is far more dynamic than that of solid-state emitters. This simultaneous tracking of molecular conformation and photophysics advances the understanding of single-molecule interactions and enables real-time sensing through two-dimensional materials.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Direct observation of individual fluorescent emitters is essential for studying quantum materials, chemical reactions, and biological systems. However, current single-molecule tracking methods only focuses on the localizations of molecules, overlooking molecular configuration and orientation. In this work, we introduce a high-throughput polarized single-molecule localization microscopy that simultaneously resolves the locations and emission dipole orientations of single fluorescent emitters with nanometer precision. Using the interface between pristine hexagonal boron nitride (h-BN) and an organic solvent as a challenging platform, we capture over 10⁵ fluorescent events and reveal distinct molecular interaction dynamics at room temperature. The measured dipole orientations align with the three-fold (C₃) rotational symmetry of the h-BN lattice, and molecular dynamics in the liquid environment can be modulated electrochemically, suggesting a route for on-demand control of quantum emitters. We also find that lateral diffusion at the solid–liquid interface is far more dynamic than that of solid-state emitters. This simultaneous tracking of molecular conformation and photophysics advances the understanding of single-molecule interactions and enables real-time sensing through two-dimensional materials. |
| 321. | | Yangning Zhang, Hyeong Woo Ban, Pan Xia, Yang Bai, Sile Hu, Filip Dinic, Chang Liu, Yixi Li, Ehsan Nikbin, Shea Sanvordenker, Muhammad Imran, Benjamin Rehl, Lewei Zeng, Sjoerd Hoogland, Xiao Qi, Brian A. Korgel, Emory Chan, Edward H. Sargent Automated synthesis of InSb quantum dots with improved batch-to-batch reproducibility via kinetically matched co-reduction In: Nature Communications, 2026, (Article in Press). @article{nokey,
title = {Automated synthesis of InSb quantum dots with improved batch-to-batch reproducibility via kinetically matched co-reduction},
author = {Yangning Zhang and Hyeong Woo Ban and Pan Xia and Yang Bai and Sile Hu and Filip Dinic and Chang Liu and Yixi Li and Ehsan Nikbin and Shea Sanvordenker and Muhammad Imran and Benjamin Rehl and Lewei Zeng and Sjoerd Hoogland and Xiao Qi and Brian A. Korgel and Emory Chan and Edward H. Sargent },
url = {https://www.nature.com/articles/s41467-026-74136-3_reference.pdf},
doi = {10.1038/s41467-026-74136-3},
year = {2026},
date = {2026-06-08},
journal = {Nature Communications},
abstract = {Indium antimonide (InSb) colloidal quantum dots (CQDs) are attractive heavy-metal-free absorbers for infrared photodetection, yet their synthesis remains challenging because precursor reduction overlaps with nucleation and growth, hindering kinetic control and yielding broad size distributions. Here we employ an automated workflow to achieve precise control over InSb CQD synthesis, leading to improved batch-to-batch reproducibility and narrow size distributions without laborious post-synthetic size-selective precipitation. We find that InSb CQD formation proceeds through a kinetically matched precursor co-reduction pathway, which requires an In-rich environment to compensate for the faster reduction of Sb3+ precursor. Within this framework, we tune CQD size by modulating precursor conversion kinetics through In/Sb precursor molar ratio and reducing agent availability. This kinetically guided size control tunes the first excitonic absorption peak of CQDs across 1120-1650 nm in the short-wave infrared. Optimized CQDs with 0.825 eV bandgap exhibit a small Stokes shift of 32 meV, which is among the smallest reported for InSb CQDs.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Indium antimonide (InSb) colloidal quantum dots (CQDs) are attractive heavy-metal-free absorbers for infrared photodetection, yet their synthesis remains challenging because precursor reduction overlaps with nucleation and growth, hindering kinetic control and yielding broad size distributions. Here we employ an automated workflow to achieve precise control over InSb CQD synthesis, leading to improved batch-to-batch reproducibility and narrow size distributions without laborious post-synthetic size-selective precipitation. We find that InSb CQD formation proceeds through a kinetically matched precursor co-reduction pathway, which requires an In-rich environment to compensate for the faster reduction of Sb3+ precursor. Within this framework, we tune CQD size by modulating precursor conversion kinetics through In/Sb precursor molar ratio and reducing agent availability. This kinetically guided size control tunes the first excitonic absorption peak of CQDs across 1120-1650 nm in the short-wave infrared. Optimized CQDs with 0.825 eV bandgap exhibit a small Stokes shift of 32 meV, which is among the smallest reported for InSb CQDs. |
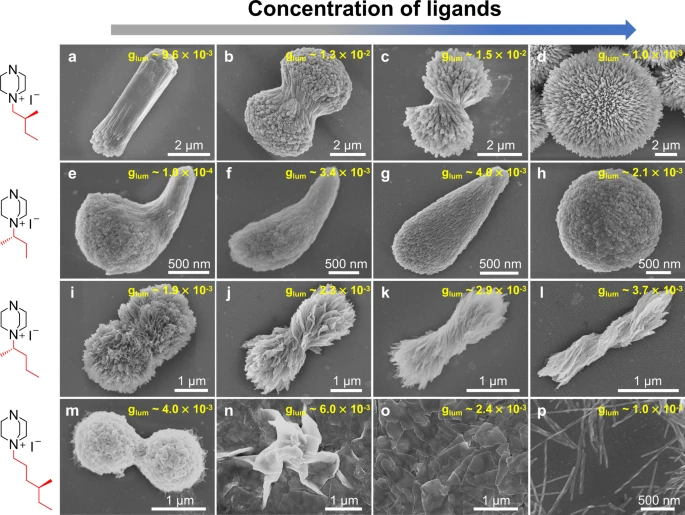
| 320. | | Haifeng Sun, Yilun Zhang, Xiao Chen, Wentao Wang, Guang-Jie Xia, Zhifeng Huang Multimodal deep-learning optimization of chiroptical properties in all-inorganic perovskite-coated TiO2 nanohelices and inverse-design transfer to organic chiral luminophores In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Multimodal deep-learning optimization of chiroptical properties in all-inorganic perovskite-coated TiO2 nanohelices and inverse-design transfer to organic chiral luminophores},
author = {Haifeng Sun and Yilun Zhang and Xiao Chen and Wentao Wang and Guang-Jie Xia and Zhifeng Huang },
url = {https://www.nature.com/articles/s41467-026-74010-2_reference.pdf},
doi = {10.1038/s41467-026-74010-2},
year = {2026},
date = {2026-06-04},
journal = {Nature Communications},
volume = {17},
abstract = {Circularly polarized luminescence (CPL) has been catching increasing attention for developing advanced photonic displays, quantum communication, bioimaging, and chiral sensing. All-inorganic chiral luminophores are superior to their organic or organic-inorganic hybrid counterparts in thermal stability, environmental robustness and device compatibility, but limited by the difficulty in fabrication and low luminescence dissymmetry factor (glum < 0.1), whereby glum is generally applied to evaluate the purity of circular polarization of CPL. Herein, chiral TiO2 nanohelices (NHs) act as chiral templates that are conformally coated with achiral perovskite luminophores composed of cesium lead bromides, to form all-inorganic chiral core@shell nano-luminophores. Chirality transmission from TiO2 NHs to perovskites accounts for the generation of CPL. Given by the complex and multifactorial experimental conditions, the manual engineering of fabrication procedure leads to an optimized glum = 0.2. To further optimize glum, we develop OptiCPL, a few-shot multimodal deep-learning framework that integrates spectral and morphological features, to boost glum from 0.20 to 0.35 through model prediction and experimental validation. In addition, the OptiCPL model is transferrable to polymer F8BT-based chiral organic luminophores, achieving glum = 0.87. This work establishes a synergistic chiral core@shell approach and offers a transferable deep-learning framework for designing high-glum CPL materials.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Circularly polarized luminescence (CPL) has been catching increasing attention for developing advanced photonic displays, quantum communication, bioimaging, and chiral sensing. All-inorganic chiral luminophores are superior to their organic or organic-inorganic hybrid counterparts in thermal stability, environmental robustness and device compatibility, but limited by the difficulty in fabrication and low luminescence dissymmetry factor (glum < 0.1), whereby glum is generally applied to evaluate the purity of circular polarization of CPL. Herein, chiral TiO2 nanohelices (NHs) act as chiral templates that are conformally coated with achiral perovskite luminophores composed of cesium lead bromides, to form all-inorganic chiral core@shell nano-luminophores. Chirality transmission from TiO2 NHs to perovskites accounts for the generation of CPL. Given by the complex and multifactorial experimental conditions, the manual engineering of fabrication procedure leads to an optimized glum = 0.2. To further optimize glum, we develop OptiCPL, a few-shot multimodal deep-learning framework that integrates spectral and morphological features, to boost glum from 0.20 to 0.35 through model prediction and experimental validation. In addition, the OptiCPL model is transferrable to polymer F8BT-based chiral organic luminophores, achieving glum = 0.87. This work establishes a synergistic chiral core@shell approach and offers a transferable deep-learning framework for designing high-glum CPL materials. |
| 319. | | Yuehua Zhao, Hongbo Chen, Ming Hu, Wen-Sheng Xu, Dapeng Wang Diffusional aging at water/oil interfaces laden with charged nanoparticles studied by single-molecule tracking In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Diffusional aging at water/oil interfaces laden with charged nanoparticles studied by single-molecule tracking},
author = {Yuehua Zhao and Hongbo Chen and Ming Hu and Wen-Sheng Xu and Dapeng Wang },
url = {https://www.nature.com/articles/s41467-026-74008-w_reference.pdf},
doi = {10.1038/s41467-026-74008-w},
year = {2026},
date = {2026-06-03},
journal = {Nature Communications},
volume = {17},
abstract = {The adsorption of charged nanoparticles at water-oil interfaces constitutes a fundamental phenomenon, underlying pivotal technologies spanning from emulsion stabilization to the sophisticated fabrication of foams. However, the diffusional behavior of these nanoparticles remains poorly understood. Here, we use single-molecule tracking experiments to show that the diffusion of like-charged nanoparticles at the water-oil interface not only becomes anomalous but also displays a diffusional aging phenomenon at the interfacial coverage that is not associated with the glassy state. We further develop a theoretical framework that quantitatively reproduces all experimental observations. Molecular dynamics simulations reveal the necessity of the coexistence of attraction and repulsion for the emergence of aging dynamics. The interplay between attraction and repulsion leads to nanoparticle adhesion taking place over observable timescales, which is manifested as aging dynamics. The discovery of diffusional aging demonstrates that interfacial evolution persists beyond adsorption equilibrium, suggesting that this effect must be accounted for in applications involving water-oil interfaces laden with charged nanoparticles.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The adsorption of charged nanoparticles at water-oil interfaces constitutes a fundamental phenomenon, underlying pivotal technologies spanning from emulsion stabilization to the sophisticated fabrication of foams. However, the diffusional behavior of these nanoparticles remains poorly understood. Here, we use single-molecule tracking experiments to show that the diffusion of like-charged nanoparticles at the water-oil interface not only becomes anomalous but also displays a diffusional aging phenomenon at the interfacial coverage that is not associated with the glassy state. We further develop a theoretical framework that quantitatively reproduces all experimental observations. Molecular dynamics simulations reveal the necessity of the coexistence of attraction and repulsion for the emergence of aging dynamics. The interplay between attraction and repulsion leads to nanoparticle adhesion taking place over observable timescales, which is manifested as aging dynamics. The discovery of diffusional aging demonstrates that interfacial evolution persists beyond adsorption equilibrium, suggesting that this effect must be accounted for in applications involving water-oil interfaces laden with charged nanoparticles. |
| 318. | | Johanna I. Hütner-Reisch, Andrea Conti, David Kugler, Florian Mittendorfer, Michael Schmid, Ulrike Diebold, Jan Balajka AFM imaging reveals the unreconstructed α‑Al2O3(0001) surface to be inhomogeneous and rough In: Nature Communications, vol. 17, no. 4692, 2026, (The unreconstructed α-Al2O3(0001) surface is widely assumed to be atomically flat. Here, the authors use noncontact-AFM to show that it is rough and laterally inhomogeneous, challenging structural models used for alumina surfaces across materials science.). @article{nokey,
title = {AFM imaging reveals the unreconstructed α‑Al2O3(0001) surface to be inhomogeneous and rough},
author = {Johanna I. Hütner-Reisch and Andrea Conti and David Kugler and Florian Mittendorfer and Michael Schmid and Ulrike Diebold and Jan Balajka },
url = {https://www.nature.com/articles/s41467-026-73690-0.pdf},
doi = {10.1038/s41467-026-73690-0},
year = {2026},
date = {2026-05-27},
urldate = {2026-05-27},
journal = {Nature Communications},
volume = {17},
number = {4692},
abstract = {Alumina (Al2O3) is a key material for thin-film growth and heterogeneous catalysis, where the atomic surface structure critically impacts performance. Using noncontact atomic force microscopy (nc-AFM) combined with density functional theory (DFT) calculations, we challenge the common assumption that the unreconstructed α-Al2O3(0001) surface is atomically flat and uniformly Al-terminated. This widely accepted bulk termination satisfies polarity compensation requirements but results in highly undercoordinated Al cations at the surface. Despite substantial inward relaxation of these Al cations, we find that the (1 × 1) surface remains inherently metastable, relative to the thermodynamically stable
R ± 9° surface reconstruction that forms at high temperatures above 1000 °C. Nc-AFM imaging of the unreconstructed surface reveals a rough and disordered morphology, with only nanometer-scale regions exhibiting the ordered Al-terminated (1 × 1) structure. Our results show that the unreconstructed Al2O3(0001) surface is intrinsically inhomogeneous, reconciling conflicting experimental observations and challenging the validity of commonly used atomistic models.},
note = {The unreconstructed α-Al2O3(0001) surface is widely assumed to be atomically flat. Here, the authors use noncontact-AFM to show that it is rough and laterally inhomogeneous, challenging structural models used for alumina surfaces across materials science.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Alumina (Al2O3) is a key material for thin-film growth and heterogeneous catalysis, where the atomic surface structure critically impacts performance. Using noncontact atomic force microscopy (nc-AFM) combined with density functional theory (DFT) calculations, we challenge the common assumption that the unreconstructed α-Al2O3(0001) surface is atomically flat and uniformly Al-terminated. This widely accepted bulk termination satisfies polarity compensation requirements but results in highly undercoordinated Al cations at the surface. Despite substantial inward relaxation of these Al cations, we find that the (1 × 1) surface remains inherently metastable, relative to the thermodynamically stable
R ± 9° surface reconstruction that forms at high temperatures above 1000 °C. Nc-AFM imaging of the unreconstructed surface reveals a rough and disordered morphology, with only nanometer-scale regions exhibiting the ordered Al-terminated (1 × 1) structure. Our results show that the unreconstructed Al2O3(0001) surface is intrinsically inhomogeneous, reconciling conflicting experimental observations and challenging the validity of commonly used atomistic models. |
| 317. | | Lexin Ding, Eduard Matito, Christian Schilling Chemical bonding concepts emerge naturally from maximally entangled atomic orbitals In: Nature Communications, vol. 17, no. 4732, 2026, (Chemical bonding explains how atoms bind together, but it remains hard to define in universal terms. Here, the authors use quantum entanglement to uncover and quantify bonds in complex systems beyond the standard Lewis paradigm.). @article{nokey,
title = {Chemical bonding concepts emerge naturally from maximally entangled atomic orbitals},
author = {Lexin Ding and Eduard Matito and Christian Schilling },
url = {https://www.nature.com/articles/s41467-026-73527-w.pdf},
doi = {10.1038/s41467-026-73527-w},
year = {2026},
date = {2026-05-27},
journal = {Nature Communications},
volume = {17},
number = {4732},
abstract = {Chemical bonding is a nonlocal phenomenon that binds atoms into molecules. Its ubiquitous presence in chemistry, however, stands in stark contrast to its ambiguous definition and the lack of a universal perspective for its understanding. In this work, we rationalize and characterize chemical bonding through the lens of an equally nonlocal concept from quantum information, the orbital entanglement. We introduce the maximally entangled atomic orbitals (MEAOs) whose entanglement pattern is shown to recover both Lewis (two-center) and beyond-Lewis (multicenter) structures, with multipartite entanglement serving as a comprehensive index of bond strength. Our unifying framework for bonding analyses is effective not only for equilibrium geometries but also for transition states in chemical reactions and complex phenomena such as aromaticity. It also has the potential to elevate the Hilbert space atomic partitioning to match the prevalent real-space partitioning in the theory of atoms in molecules. Accordingly, our work provides a new framework for understanding fuzzy chemical concepts using rigorous, quantitative descriptors from quantum information.},
note = {Chemical bonding explains how atoms bind together, but it remains hard to define in universal terms. Here, the authors use quantum entanglement to uncover and quantify bonds in complex systems beyond the standard Lewis paradigm.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chemical bonding is a nonlocal phenomenon that binds atoms into molecules. Its ubiquitous presence in chemistry, however, stands in stark contrast to its ambiguous definition and the lack of a universal perspective for its understanding. In this work, we rationalize and characterize chemical bonding through the lens of an equally nonlocal concept from quantum information, the orbital entanglement. We introduce the maximally entangled atomic orbitals (MEAOs) whose entanglement pattern is shown to recover both Lewis (two-center) and beyond-Lewis (multicenter) structures, with multipartite entanglement serving as a comprehensive index of bond strength. Our unifying framework for bonding analyses is effective not only for equilibrium geometries but also for transition states in chemical reactions and complex phenomena such as aromaticity. It also has the potential to elevate the Hilbert space atomic partitioning to match the prevalent real-space partitioning in the theory of atoms in molecules. Accordingly, our work provides a new framework for understanding fuzzy chemical concepts using rigorous, quantitative descriptors from quantum information. |
| 316. | | Miguel Ángel López Carrillo, Filip Desmet, Maksiem Erkens, Jeffrey A. Fagan, Ming Zheng, Han Li, Benjamin S. Flavel, Wim Wenseleers, Wouter Herrebout, Sofie Cambré, Dmitry I. Levshov Absolute quantification of enantiomeric purity of sorted carbon nanotubes by correlating hyperspectral fluorescence microscopy with ensemble chiroptical spectroscopy In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Absolute quantification of enantiomeric purity of sorted carbon nanotubes by correlating hyperspectral fluorescence microscopy with ensemble chiroptical spectroscopy},
author = {Miguel Ángel López Carrillo and Filip Desmet and Maksiem Erkens and Jeffrey A. Fagan and Ming Zheng and Han Li and Benjamin S. Flavel and Wim Wenseleers and Wouter Herrebout and Sofie Cambré and Dmitry I. Levshov },
url = {https://www.nature.com/articles/s41467-026-73397-2_reference.pdf},
doi = {10.1038/s41467-026-73397-2},
year = {2026},
date = {2026-05-26},
journal = {Nature Communications},
volume = {17},
abstract = {Accurate determination of enantiomeric purity is essential for advancing chiral materials in nanotechnology, optoelectronics, and quantum information science. Chiroptical spectroscopic techniques provide rapid, non-destructive measurements of enantiomeric excess (ee), but their use for complex systems like single-walled carbon nanotubes (SWCNTs) is limited by the lack of enantiopure references for calibration. Here we demonstrate an absolute approach combining hyperspectral imaging (HSI) with single-nanotube counting statistics and ensemble chiroptical spectroscopy such as electronic circular dichroism (ECD) and Raman optical activity (ROA) to quantify ee without requiring such standards. Analysis of thousands of individual nanotubes reveals sensitivity of HSI and chiroptical responses to synthesis, purification, and SWCNT concentration, highlighting pronounced source-dependent inhomogeneity. Nevertheless, universal calibration curves for ECD and ROA intensities are established from the purest, most uniform enantiomer-sorted samples. This methodology is extendable to other SWCNT chiralities and chiroptical techniques, enabling quantitative enantiomer sorting and systematic investigations of chirality-dependent properties and applications.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Accurate determination of enantiomeric purity is essential for advancing chiral materials in nanotechnology, optoelectronics, and quantum information science. Chiroptical spectroscopic techniques provide rapid, non-destructive measurements of enantiomeric excess (ee), but their use for complex systems like single-walled carbon nanotubes (SWCNTs) is limited by the lack of enantiopure references for calibration. Here we demonstrate an absolute approach combining hyperspectral imaging (HSI) with single-nanotube counting statistics and ensemble chiroptical spectroscopy such as electronic circular dichroism (ECD) and Raman optical activity (ROA) to quantify ee without requiring such standards. Analysis of thousands of individual nanotubes reveals sensitivity of HSI and chiroptical responses to synthesis, purification, and SWCNT concentration, highlighting pronounced source-dependent inhomogeneity. Nevertheless, universal calibration curves for ECD and ROA intensities are established from the purest, most uniform enantiomer-sorted samples. This methodology is extendable to other SWCNT chiralities and chiroptical techniques, enabling quantitative enantiomer sorting and systematic investigations of chirality-dependent properties and applications. |
| 315. | | Shilu Zhu, Shuwei Shen, Min Ye, Yang Zhang, Zhiyuan Zheng, Jie Gao, Ru Zhang, Zhongliang Lang, Peng Yao, Mingzhai Sun, Luke P. Lee, Ronald X. Xu Transport of enzymatic activity across liquid-liquid interfaces using dynamic assemblies of magnetic particles via field-modulated interactions In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Transport of enzymatic activity across liquid-liquid interfaces using dynamic assemblies of magnetic particles via field-modulated interactions},
author = {Shilu Zhu and Shuwei Shen and Min Ye and Yang Zhang and Zhiyuan Zheng and Jie Gao and Ru Zhang and Zhongliang Lang and Peng Yao and Mingzhai Sun and Luke P. Lee and Ronald X. Xu },
url = {https://www.nature.com/articles/s41467-026-73696-8_reference.pdf},
doi = {10.1038/s41467-026-73696-8},
year = {2026},
date = {2026-05-26},
journal = {Nature Communications},
volume = {17},
abstract = {Biological systems dynamically grow high-aspect-ratio architectures from a site, enabling traversal of phase boundaries and functional execution. Emulating this growth strategy in synthetic systems could yield functional microsystems for operation across interfaces. However, engineering such bio-inspired growth to proceed out of plane from a substrate in synthetic colloidal assemblies remains challenging, as it requires overcoming gravitational collapse while maintaining structural coherence during extension. Here, we present a field-driven particle system that achieves gravity-resisting growth of high-aspect-ratio structures via frequency-modulated magnetic and hydrodynamic interactions. This growth is enabled by combining static and oscillating magnetic fields, which guide the assembly of magnetic particles into dynamic structures exhibiting a distinct segmented, seaweed-like morphology. These architectures are reconfigurable, stabilizable, programmably actuatable, and capable of penetrating a perfluorohexane–water interface. When functionalized with enzymes, the growing structures act as micro-transporters, delivering catalytic activity across the interface and triggering detectable reactions in both bulk two-phase and microfluidic chip systems. This work establishes a field-driven assembly-to-function approach that integrates structural growth, phase-boundary penetration, and triggered functionality, enabling active microsystems capable of interfacial transport and functional execution.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Biological systems dynamically grow high-aspect-ratio architectures from a site, enabling traversal of phase boundaries and functional execution. Emulating this growth strategy in synthetic systems could yield functional microsystems for operation across interfaces. However, engineering such bio-inspired growth to proceed out of plane from a substrate in synthetic colloidal assemblies remains challenging, as it requires overcoming gravitational collapse while maintaining structural coherence during extension. Here, we present a field-driven particle system that achieves gravity-resisting growth of high-aspect-ratio structures via frequency-modulated magnetic and hydrodynamic interactions. This growth is enabled by combining static and oscillating magnetic fields, which guide the assembly of magnetic particles into dynamic structures exhibiting a distinct segmented, seaweed-like morphology. These architectures are reconfigurable, stabilizable, programmably actuatable, and capable of penetrating a perfluorohexane–water interface. When functionalized with enzymes, the growing structures act as micro-transporters, delivering catalytic activity across the interface and triggering detectable reactions in both bulk two-phase and microfluidic chip systems. This work establishes a field-driven assembly-to-function approach that integrates structural growth, phase-boundary penetration, and triggered functionality, enabling active microsystems capable of interfacial transport and functional execution. |
| 314. | | Yu Ren, Fu-zhi Dai, Sijia Lu, Yunfeng Hu, Qi Ding, Mingming Si, Yongsheng Sun, Zhiguo Xia, Yuchi Fan, Wan Jiang Cold sintering of hybrid copper-iodides in transparent ceramics using dolomitization-inspired densification In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Cold sintering of hybrid copper-iodides in transparent ceramics using dolomitization-inspired densification},
author = {Yu Ren and Fu-zhi Dai and Sijia Lu and Yunfeng Hu and Qi Ding and Mingming Si and Yongsheng Sun and Zhiguo Xia and Yuchi Fan and Wan Jiang },
url = {https://www.nature.com/articles/s41467-026-73625-9_reference.pdf},
doi = {10.1038/s41467-026-73625-9},
year = {2026},
date = {2026-05-25},
journal = {Nature Communications},
volume = {17},
abstract = {The non-toxic hybrid luminescent copper(I) iodides are competitive solution for environmentally sustainable applications in solar cells, lighting, and detectors. However, their susceptibility to elevated temperature and oxidation creates great hurdles for technological integration due to the lack of suitable encapsulation. Inspired by the mechanism and kinetics of dolomitization in geology, we propose a cold sintering process that enables the densification of highly transparent SrF2 ceramic at 150 oC, which can serve as a matrix for hybrid copper iodides. The introduction of cation-rich (Sr2+ rich) solution accelerates the rate-determining precipitation of solvated ions, which largely promotes the cold sintering process at low temperature. During the proposed cold sintering process, the synergistic effect of uniaxial pressure and temperature generates a continuous dispersion of melted hybrid copper iodide ([Cu4I4(pph2Et)4]) throughout the thermally conductive matrix, leading to an external quantum efficiency of 50%, enhanced resistance to thermal quenching, and superior stability after 100 h of damp-heat test. By encapsulating various hybrid copper iodides, the strategy is applicable for not only full-spectrum white lighting, but also X-ray imaging.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The non-toxic hybrid luminescent copper(I) iodides are competitive solution for environmentally sustainable applications in solar cells, lighting, and detectors. However, their susceptibility to elevated temperature and oxidation creates great hurdles for technological integration due to the lack of suitable encapsulation. Inspired by the mechanism and kinetics of dolomitization in geology, we propose a cold sintering process that enables the densification of highly transparent SrF2 ceramic at 150 oC, which can serve as a matrix for hybrid copper iodides. The introduction of cation-rich (Sr2+ rich) solution accelerates the rate-determining precipitation of solvated ions, which largely promotes the cold sintering process at low temperature. During the proposed cold sintering process, the synergistic effect of uniaxial pressure and temperature generates a continuous dispersion of melted hybrid copper iodide ([Cu4I4(pph2Et)4]) throughout the thermally conductive matrix, leading to an external quantum efficiency of 50%, enhanced resistance to thermal quenching, and superior stability after 100 h of damp-heat test. By encapsulating various hybrid copper iodides, the strategy is applicable for not only full-spectrum white lighting, but also X-ray imaging. |
| 313. | | Baowei Zhang, Kexin Chen, Zhengkun Xie, Kun Hu, Haiyun Dong, Yong Sheng Zhao, Liberato Manna, Siyu Lu One-dimensional CsPbBr3 superlattices with polarized and amplified spontaneous circularly polarized emissions In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {One-dimensional CsPbBr3 superlattices with polarized and amplified spontaneous circularly polarized emissions},
author = {Baowei Zhang and Kexin Chen and Zhengkun Xie and Kun Hu and Haiyun Dong and Yong Sheng Zhao and Liberato Manna and Siyu Lu },
url = {https://www.nature.com/articles/s41467-026-73513-2_reference.pdf},
doi = {10.1038/s41467-026-73513-2},
year = {2026},
date = {2026-05-23},
journal = {Nature Communications},
volume = {17},
abstract = {Nanocrystal superlattices typically occur in two- or three-dimensional configurations, constrained to the micrometer scale and with limited size tunability. Here, we report one-dimensional superlattices prepared by self-assembly of CsPbBr3 nanorods and nanoplatelets. These exhibit a hierarchical structure, evolving from ribbons (µm) of nanorods or nanoplatelets to assemblies of ribbons (mm). These superlattices have a diameter of 0.8–1 µm and length in the range of 13–1500 µm, and their aspect ratio can be tuned from 14 to 1200 by adjusting the shape of the nanorods and nanoplatelets. Thanks to their anisotropic structure, the superlattices show strong polarized emission with a near-unity degree of polarization, ≈4 times larger than that of randomly assembled film. These superlattices also exhibit chiral optical response (circular dichroism and circularly polarized emission). Since no chiral ligands are used in the synthesis, the chiral signal (negative or positive) from the superlattices is random. However, the signal can be controlled after the addition of chiral ligands. The maximum dissymmetry factor of the luminescence (glum) is −0.11, and can be further amplified to −0.32 in the amplified spontaneous emission of the superlattices.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanocrystal superlattices typically occur in two- or three-dimensional configurations, constrained to the micrometer scale and with limited size tunability. Here, we report one-dimensional superlattices prepared by self-assembly of CsPbBr3 nanorods and nanoplatelets. These exhibit a hierarchical structure, evolving from ribbons (µm) of nanorods or nanoplatelets to assemblies of ribbons (mm). These superlattices have a diameter of 0.8–1 µm and length in the range of 13–1500 µm, and their aspect ratio can be tuned from 14 to 1200 by adjusting the shape of the nanorods and nanoplatelets. Thanks to their anisotropic structure, the superlattices show strong polarized emission with a near-unity degree of polarization, ≈4 times larger than that of randomly assembled film. These superlattices also exhibit chiral optical response (circular dichroism and circularly polarized emission). Since no chiral ligands are used in the synthesis, the chiral signal (negative or positive) from the superlattices is random. However, the signal can be controlled after the addition of chiral ligands. The maximum dissymmetry factor of the luminescence (glum) is −0.11, and can be further amplified to −0.32 in the amplified spontaneous emission of the superlattices. |
| 312. | | Yicheng Zeng, Xiaonan Liu, Duanyang Liu, Yuan Liu, Qingya Wang, Weiwei Chen, Jing Wei, Jian Xu, Fangze Liu, Hongbo Li Selective radial thickness growth of compositionally graded shells on colloidal quantum rods for more efficient light-emitting diodes In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Selective radial thickness growth of compositionally graded shells on colloidal quantum rods for more efficient light-emitting diodes},
author = {Yicheng Zeng and Xiaonan Liu and Duanyang Liu and Yuan Liu and Qingya Wang and Weiwei Chen and Jing Wei and Jian Xu and Fangze Liu and Hongbo Li },
url = {https://www.nature.com/articles/s41467-026-73298-4_reference.pdf},
doi = {10.1038/s41467-026-73298-4},
year = {2026},
date = {2026-05-21},
journal = {Nature Communications},
volume = {17},
abstract = {Colloidal quantum nanorods (NRs) exhibit linearly polarized emission and fast radiative recombination owing to their two-dimensional confinement, enabling high-performance light-emitting diodes (LEDs). However, due to facet-dependent growth kinetics, anisotropic shell growth often leads to preferential overgrowth along the long axis and insufficient radial (short axis) coverage. This structural imbalance weakens exciton confinement within the core and compromises the suppression of Förster resonance energy transfer. Here, we report a dual-ligand (organic phosphorus/carboxylic acids) slow-injection strategy to synthesize wurtzite CdSe/CdZnSe/ZnSeS core/shell NRs featuring compositionally graded thick shell ( ≈ 4.5 nm) with ZnSe as the dominant component. The organic phosphorus ligands create an environment of high monomer concentration (cadmium monomer concentration from 1.4% to 2% of cadmium by mass) to drive anisotropic growth, while the carboxylic acids promote isotropic growth due to their nearly equal binding energy across different crystal facets. Simultaneously epitaxial growth of compositionally graded alloy shells alleviates lattice mismatch at core/shell interfaces while reducing defect density and boosting carrier recombination efficiency. The resulting NR-LEDs achieve an external quantum efficiency of 32% for anisotropic nanocrystals. This work establishes graded thick-shell NRs as a generalizable platform for efficient, stable and polarized optoelectronics.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Colloidal quantum nanorods (NRs) exhibit linearly polarized emission and fast radiative recombination owing to their two-dimensional confinement, enabling high-performance light-emitting diodes (LEDs). However, due to facet-dependent growth kinetics, anisotropic shell growth often leads to preferential overgrowth along the long axis and insufficient radial (short axis) coverage. This structural imbalance weakens exciton confinement within the core and compromises the suppression of Förster resonance energy transfer. Here, we report a dual-ligand (organic phosphorus/carboxylic acids) slow-injection strategy to synthesize wurtzite CdSe/CdZnSe/ZnSeS core/shell NRs featuring compositionally graded thick shell ( ≈ 4.5 nm) with ZnSe as the dominant component. The organic phosphorus ligands create an environment of high monomer concentration (cadmium monomer concentration from 1.4% to 2% of cadmium by mass) to drive anisotropic growth, while the carboxylic acids promote isotropic growth due to their nearly equal binding energy across different crystal facets. Simultaneously epitaxial growth of compositionally graded alloy shells alleviates lattice mismatch at core/shell interfaces while reducing defect density and boosting carrier recombination efficiency. The resulting NR-LEDs achieve an external quantum efficiency of 32% for anisotropic nanocrystals. This work establishes graded thick-shell NRs as a generalizable platform for efficient, stable and polarized optoelectronics. |
| 311. | | Xiaoyun Guo, Yujing Zhang, Chao Yang, Yunhao Lu, Zimo Lin, Min Tang, Guanxing Li, Yang Ou, Beien Zhu, Ying Jiang, Zhong-kang Han, Wentao Yuan, Yi Gao, Ze Zhang, Yong Wang Atomic-level insights into the high intrinsic thermostability of individual anatase TiO2 nanocrystals through surface-locking effects In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Atomic-level insights into the high intrinsic thermostability of individual anatase TiO2 nanocrystals through surface-locking effects},
author = {Xiaoyun Guo and Yujing Zhang and Chao Yang and Yunhao Lu and Zimo Lin and Min Tang and Guanxing Li and Yang Ou and Beien Zhu and Ying Jiang and Zhong-kang Han and Wentao Yuan and Yi Gao and Ze Zhang and Yong Wang },
url = {https://www.nature.com/articles/s41467-026-73332-5_reference.pdf},
doi = {10.1038/s41467-026-73332-5},
year = {2026},
date = {2026-05-20},
journal = {Nature Communications},
volume = {17},
abstract = {Nanocrystal phase thermostability is critical for their applications, yet fundamentally governed by complex thermodynamic and kinetic variables. Understanding the stabilizing mechanisms and dominant factors requires atomic-level insights into dynamic evolution across surface and bulk regions under extreme conditions. Herein, we present a comprehensive in-situ investigation of individual single-crystalline anatase TiO2 nanorods using spherical aberration-corrected scanning transmission electron microscopy. By simultaneously acquiring environmental secondary electron images for surface topography and high-angle annular dark-field images for bulk atomic structures, we reveal the extraordinary phase stability of individual anatase nanorods governed by surface effects, distinct from aggregated nanorods. Anatase TiO2 nanorods undergo morphology reshaping and surface atomic reconstruction above 600 °C, involving transformation from high-index surfaces to (101) facets and the formation of (1 × 4)-reconstructed (001) surfaces. Remarkably, individual anatase TiO2 nanorods maintain the anatase structure even up to 1250 °C without transforming into the rutile phase. The restructuring lowers the total energy of the system, and acts as a kinetic “surface-locking” effect preventing rutile nucleation. Beyond elucidating the restructuring mechanisms and intrinsic thermostability of TiO2 nanocrystals, this work also establishes an effective pathway for simultaneously probing the complex structural evolution of nanomaterials across both surface and bulk regions.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanocrystal phase thermostability is critical for their applications, yet fundamentally governed by complex thermodynamic and kinetic variables. Understanding the stabilizing mechanisms and dominant factors requires atomic-level insights into dynamic evolution across surface and bulk regions under extreme conditions. Herein, we present a comprehensive in-situ investigation of individual single-crystalline anatase TiO2 nanorods using spherical aberration-corrected scanning transmission electron microscopy. By simultaneously acquiring environmental secondary electron images for surface topography and high-angle annular dark-field images for bulk atomic structures, we reveal the extraordinary phase stability of individual anatase nanorods governed by surface effects, distinct from aggregated nanorods. Anatase TiO2 nanorods undergo morphology reshaping and surface atomic reconstruction above 600 °C, involving transformation from high-index surfaces to (101) facets and the formation of (1 × 4)-reconstructed (001) surfaces. Remarkably, individual anatase TiO2 nanorods maintain the anatase structure even up to 1250 °C without transforming into the rutile phase. The restructuring lowers the total energy of the system, and acts as a kinetic “surface-locking” effect preventing rutile nucleation. Beyond elucidating the restructuring mechanisms and intrinsic thermostability of TiO2 nanocrystals, this work also establishes an effective pathway for simultaneously probing the complex structural evolution of nanomaterials across both surface and bulk regions. |
| 310. | | Zhuang Wang, Cong Fang, Lili Zhang, Wenshuai Zhu, Yuxiao Ding, Sicong Ma, Xiaoyan Sun General workflow for localizing hydrides in metal nanoclusters by combining stochastic surface walking with neural-network potentials In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {General workflow for localizing hydrides in metal nanoclusters by combining stochastic surface walking with neural-network potentials},
author = {Zhuang Wang and Cong Fang and Lili Zhang and Wenshuai Zhu and Yuxiao Ding and Sicong Ma and Xiaoyan Sun },
url = {https://www.nature.com/articles/s41467-026-72966-9_reference.pdf},
doi = {10.1038/s41467-026-72966-9},
year = {2026},
date = {2026-05-11},
urldate = {2026-05-11},
journal = {Nature Communications},
volume = {17},
abstract = {Ligand-protected metal hydride nanoclusters are crucial for applications in catalysis, luminescence, and energy technologies. However, accurately locating hydrogen atoms (hydrides) within these complex structures remains a significant challenge, hindering the full exploitation of their properties. Developing a universal and accessible method for hydride localization is essential. Here, we present a general computational workflow that combines global structural search algorithms with machine learning-based neural-network potentials to efficiently locate hydrides. We validate this approach across 93 experimentally reported systems, including coinage-metal, transition-metal, and multimetallic polyoxometalates. Using this framework, here we show the generalized rules governing hydride positioning and their preferred coordination environments. Furthermore, we reveal the atomic-level dynamics of hydride movement, discovering that surface migration is the predominant pathway. Practically, our approach provides a reliable theoretical supplement to resolve uncertainties in experimental hydride quantification, such as those from mass spectrometry. Overall, this study advances the fundamental understanding of hydride behavior in nanoclusters and offers a robust, predictive tool to guide the synthesis and structural characterization of nanomaterials.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ligand-protected metal hydride nanoclusters are crucial for applications in catalysis, luminescence, and energy technologies. However, accurately locating hydrogen atoms (hydrides) within these complex structures remains a significant challenge, hindering the full exploitation of their properties. Developing a universal and accessible method for hydride localization is essential. Here, we present a general computational workflow that combines global structural search algorithms with machine learning-based neural-network potentials to efficiently locate hydrides. We validate this approach across 93 experimentally reported systems, including coinage-metal, transition-metal, and multimetallic polyoxometalates. Using this framework, here we show the generalized rules governing hydride positioning and their preferred coordination environments. Furthermore, we reveal the atomic-level dynamics of hydride movement, discovering that surface migration is the predominant pathway. Practically, our approach provides a reliable theoretical supplement to resolve uncertainties in experimental hydride quantification, such as those from mass spectrometry. Overall, this study advances the fundamental understanding of hydride behavior in nanoclusters and offers a robust, predictive tool to guide the synthesis and structural characterization of nanomaterials. |
| 309. | | Qinwei An, Xueyang Zhang, Yuanlin Fang, Wen Zhao, Wenqi Xiong, Feng Li, Shengjun Yuan, Jie Wang Atomic dynamics of solid-gas interfaces unveil dual-layer formation and WS2 nucleation driven by multistep phase-transition In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Atomic dynamics of solid-gas interfaces unveil dual-layer formation and WS2 nucleation driven by multistep phase-transition},
author = {Qinwei An and Xueyang Zhang and Yuanlin Fang and Wen Zhao and Wenqi Xiong and Feng Li and Shengjun Yuan and Jie Wang },
url = {https://www.nature.com/articles/s41467-026-72731-y_reference.pdf},
doi = {10.1038/s41467-026-72731-y},
year = {2026},
date = {2026-05-08},
journal = {Nature Communications},
volume = {17},
abstract = {Atomic-scale solid–gas interface (SGI) dynamics remain elusive due to transient intermediates, complex interfacial environments, and challenges of real-time characterization. Using an environmental transmission electron microscopy cell as a microreaction chamber combined with atomic-resolution in-situ imaging, here we directly visualize SGI reactions at the interface of transition metal oxidate WO2.72 nanowire under reactive gas environments. We reveal that initial SGI interactions trigger surface restructuring into a dual-layer configuration, consisting of an uppermost amorphous layer and an underlying lattice-distorted condensed region. The amorphous surface layer acts as a quasi-liquid precursor reservoir that promotes reversible crystalline–amorphous transformations and short-range ordering for critical nucleus formation, while the roughened, defect-rich subsurface interface provides energetically favorable sites for WS2 nucleation and vertical growth. Furthermore, in-situ atomic-scale observations of MoS2 nucleation and growth via SGI reactions demonstrate the generality of this mechanism. The atomistic processes governing interfacial reconstruction and nucleation are further corroborated by theoretical calculations. Our results establish a dual-layer-mediated reconstruction pathway during SGI reactions, overturning the conventional view of atomically sharp and static reaction fronts. Moreover, these findings provide insights into multistep phase-transition-governed WS2 nucleation and growth, enabling controlled synthesis of 2D WS2 and MoS2 toward atomic-scale manufacturing.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Atomic-scale solid–gas interface (SGI) dynamics remain elusive due to transient intermediates, complex interfacial environments, and challenges of real-time characterization. Using an environmental transmission electron microscopy cell as a microreaction chamber combined with atomic-resolution in-situ imaging, here we directly visualize SGI reactions at the interface of transition metal oxidate WO2.72 nanowire under reactive gas environments. We reveal that initial SGI interactions trigger surface restructuring into a dual-layer configuration, consisting of an uppermost amorphous layer and an underlying lattice-distorted condensed region. The amorphous surface layer acts as a quasi-liquid precursor reservoir that promotes reversible crystalline–amorphous transformations and short-range ordering for critical nucleus formation, while the roughened, defect-rich subsurface interface provides energetically favorable sites for WS2 nucleation and vertical growth. Furthermore, in-situ atomic-scale observations of MoS2 nucleation and growth via SGI reactions demonstrate the generality of this mechanism. The atomistic processes governing interfacial reconstruction and nucleation are further corroborated by theoretical calculations. Our results establish a dual-layer-mediated reconstruction pathway during SGI reactions, overturning the conventional view of atomically sharp and static reaction fronts. Moreover, these findings provide insights into multistep phase-transition-governed WS2 nucleation and growth, enabling controlled synthesis of 2D WS2 and MoS2 toward atomic-scale manufacturing. |
| 308. | | Junbin Li, Fernando Delgado-Licona, Zhenyang Liu, Hayden Perry, Jinge Xu, Nikolai Mukhin, Sina Sadeghi, Ou Chen, Milad Abolhasani Autonomous microfluidic experimentation for exploring reaction inference and synthesizing double perovskite nanoplatelets In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Autonomous microfluidic experimentation for exploring reaction inference and synthesizing double perovskite nanoplatelets},
author = {Junbin Li and Fernando Delgado-Licona and Zhenyang Liu and Hayden Perry and Jinge Xu and Nikolai Mukhin and Sina Sadeghi and Ou Chen and Milad Abolhasani },
url = {https://www.nature.com/articles/s41467-026-72765-2_reference.pdf},
doi = {10.1038/s41467-026-72765-2},
year = {2026},
date = {2026-05-04},
urldate = {2026-05-04},
journal = {Nature Communications},
volume = {17},
abstract = {Self-driving laboratories enable accelerated exploration of chemical and materials spaces by coupling automated experimentation with machine-learning-guided decision making. However, extending autonomous discovery to compositionally complex materials with multiple coupled reaction pathways remains a significant challenge. Here, we introduce PoLARIS, a microfluidic self-driving laboratory designed for time- and material-efficient autonomous synthesis, optimization, and mechanistic interrogation of multi-element nanocrystals. Using PoLARIS, we achieve rapid data-driven optimization of metal halide double perovskite nanoplatelets, comprising up to six distinct elements synthesized via a continuous-flow heat-up reaction. The platform integrates a modular microfluidic reactor architecture with closed-loop experiment selection to efficiently navigate a high-dimensional synthesis parameter space. Beyond autonomous multi-element nanoplatelet synthesis and optimization, PoLARIS utilizes dynamic flow experimentation to enable mechanistic inference of precursor reactivity and reaction pathways governing nanoplatelet formation. This work establishes microfluidic self-driving laboratories as a generalizable approach for unifying autonomous synthesis optimization with mechanistic understanding in compositionally complex colloidal materials systems. PoLARIS framework provides a scalable pathway toward autonomous discovery in other multi-element and high-entropy colloidal nanocrystals beyond double perovskites.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Self-driving laboratories enable accelerated exploration of chemical and materials spaces by coupling automated experimentation with machine-learning-guided decision making. However, extending autonomous discovery to compositionally complex materials with multiple coupled reaction pathways remains a significant challenge. Here, we introduce PoLARIS, a microfluidic self-driving laboratory designed for time- and material-efficient autonomous synthesis, optimization, and mechanistic interrogation of multi-element nanocrystals. Using PoLARIS, we achieve rapid data-driven optimization of metal halide double perovskite nanoplatelets, comprising up to six distinct elements synthesized via a continuous-flow heat-up reaction. The platform integrates a modular microfluidic reactor architecture with closed-loop experiment selection to efficiently navigate a high-dimensional synthesis parameter space. Beyond autonomous multi-element nanoplatelet synthesis and optimization, PoLARIS utilizes dynamic flow experimentation to enable mechanistic inference of precursor reactivity and reaction pathways governing nanoplatelet formation. This work establishes microfluidic self-driving laboratories as a generalizable approach for unifying autonomous synthesis optimization with mechanistic understanding in compositionally complex colloidal materials systems. PoLARIS framework provides a scalable pathway toward autonomous discovery in other multi-element and high-entropy colloidal nanocrystals beyond double perovskites. |
| 307. | | Xiulin Fan, Chen Zhang, Mengtian Chen, Qin Zhang, Lin Wei, Zhongju Ye, Lehui Xiao Single-particle catalytic monitoring of interfacial charge dynamics during light-driven CdS shell growth on Au nanocubes In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Single-particle catalytic monitoring of interfacial charge dynamics during light-driven CdS shell growth on Au nanocubes},
author = {Xiulin Fan and Chen Zhang and Mengtian Chen and Qin Zhang and Lin Wei and Zhongju Ye and Lehui Xiao },
url = {https://www.nature.com/articles/s41467-026-71517-6_reference.pdf},
doi = {10.1038/s41467-026-71517-6},
year = {2026},
date = {2026-05-04},
journal = {Nature Communications},
volume = {17},
abstract = {Noble metal nanomaterials, with localized surface plasmon resonance (LSPR) properties, have shown great promise in photocatalysis. However, understanding plasmon-induced interfacial charge transfer dynamics and optimizing photocatalytic efficiency remain significant challenges. Here we show the precise control of the growth of CdS shells through light-driven deposition while monitoring interfacial charge dynamics by using Au nanocubes (Au NCs) as plasmonic substrates. We discover a non-monotonic relationship between the CdS shell thickness and photocatalytic activity. An optimal shell thickness maximizes the hot-electron transfer efficiency, achieving a 2.62-fold enhancement in catalytic activity compared to bare Au NCs by effectively balancing carrier separation and recombination. Single-particle analysis, through synchronized dark-field scattering and single-molecule catalytic imaging, reveals heterogeneous catalytic behaviors associated with atomic-scale structural variations and dynamic charge transfer mechanisms. The localized electromagnetic field enhancement around Au NCs facilitates hot-carrier generation for CdS deposition, and interfacial Schottky barriers extend hot-carrier lifetimes for redox reactions. This work provides a framework for designing dynamic metal-semiconductor interfaces. It not only deepens our fundamental understanding of plasmon-induced energy conversion but also serves as a guiding principle for the development of high-efficiency solar fuel systems.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Noble metal nanomaterials, with localized surface plasmon resonance (LSPR) properties, have shown great promise in photocatalysis. However, understanding plasmon-induced interfacial charge transfer dynamics and optimizing photocatalytic efficiency remain significant challenges. Here we show the precise control of the growth of CdS shells through light-driven deposition while monitoring interfacial charge dynamics by using Au nanocubes (Au NCs) as plasmonic substrates. We discover a non-monotonic relationship between the CdS shell thickness and photocatalytic activity. An optimal shell thickness maximizes the hot-electron transfer efficiency, achieving a 2.62-fold enhancement in catalytic activity compared to bare Au NCs by effectively balancing carrier separation and recombination. Single-particle analysis, through synchronized dark-field scattering and single-molecule catalytic imaging, reveals heterogeneous catalytic behaviors associated with atomic-scale structural variations and dynamic charge transfer mechanisms. The localized electromagnetic field enhancement around Au NCs facilitates hot-carrier generation for CdS deposition, and interfacial Schottky barriers extend hot-carrier lifetimes for redox reactions. This work provides a framework for designing dynamic metal-semiconductor interfaces. It not only deepens our fundamental understanding of plasmon-induced energy conversion but also serves as a guiding principle for the development of high-efficiency solar fuel systems. |
| 306. | | Xiaoxi Luan, Fengxia Wu, Yu Tian, Fenghua Li, Wenping Gao, Guobao Xu, Guangchao Zheng, Wing-Leung Wong, Kwok-Yin Wong, Wenxin Niu Structural origin of hierarchical chirality in Pt@Au nanostructures with step-rich surfaces In: Nature Communications, 2026, (Article in Press). @article{nokey,
title = {Structural origin of hierarchical chirality in Pt@Au nanostructures with step-rich surfaces},
author = {Xiaoxi Luan and Fengxia Wu and Yu Tian and Fenghua Li and Wenping Gao and Guobao Xu and Guangchao Zheng and Wing-Leung Wong and Kwok-Yin Wong and Wenxin Niu },
url = {https://www.nature.com/articles/s41467-026-72761-6_reference.pdf},
doi = {10.1038/s41467-026-72761-6},
year = {2026},
date = {2026-04-30},
journal = {Nature Communications},
abstract = {Hierarchical chirality, arising from the coupling of chiral surfaces, units, assemblies, and overall architectures, represents a structural paradigm with profound implications for enantioselective catalysis, molecular recognition, and chiral photonics. Unraveling and regulating its formation across multiple length scales is crucial for understanding structure–property relationships and guiding the design of chiral nanomaterials. Here, we present a strategy for constructing step-rich Pt@Au nanostructures with multilevel chirality. Using octahedral Pt nanoframes as templates and cysteine as a chiral inducer, rotated and oriented Au triangular pyramids with high-Miller-index chiral facets are generated, leading to the formation of fishbone-like, pinwheel-like, and tilted-step surface features that collectively constitute a hierarchical chiral nanostructure. Mechanistic studies identify cysteine and Pt nanoframes as indispensable for directing step formation and hierarchical growth, thereby defining the overall morphology and chiroplasmonic responses. Leveraging the abundant chiral surface area and the strong chiral electromagnetic nearfields generated under plasmonic excitation, we fabricate monolayer-modified electrodes based on the Pt@Au nanostructures that enabled plasmon-enhanced enantioselective electrochemical recognition of penicillamine enantiomers and determination of their enantiomeric purity. This strategy clarifies the origin of multilevel chirality in step-rich architectures and establishes a structural engineering approach for designing chiral nanomaterials with enhanced near-field properties for enantioselective recognition.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hierarchical chirality, arising from the coupling of chiral surfaces, units, assemblies, and overall architectures, represents a structural paradigm with profound implications for enantioselective catalysis, molecular recognition, and chiral photonics. Unraveling and regulating its formation across multiple length scales is crucial for understanding structure–property relationships and guiding the design of chiral nanomaterials. Here, we present a strategy for constructing step-rich Pt@Au nanostructures with multilevel chirality. Using octahedral Pt nanoframes as templates and cysteine as a chiral inducer, rotated and oriented Au triangular pyramids with high-Miller-index chiral facets are generated, leading to the formation of fishbone-like, pinwheel-like, and tilted-step surface features that collectively constitute a hierarchical chiral nanostructure. Mechanistic studies identify cysteine and Pt nanoframes as indispensable for directing step formation and hierarchical growth, thereby defining the overall morphology and chiroplasmonic responses. Leveraging the abundant chiral surface area and the strong chiral electromagnetic nearfields generated under plasmonic excitation, we fabricate monolayer-modified electrodes based on the Pt@Au nanostructures that enabled plasmon-enhanced enantioselective electrochemical recognition of penicillamine enantiomers and determination of their enantiomeric purity. This strategy clarifies the origin of multilevel chirality in step-rich architectures and establishes a structural engineering approach for designing chiral nanomaterials with enhanced near-field properties for enantioselective recognition. |
| 305. | | Robin E. Bumbaugh, Doran L. Pennington, Lena C. Wehn, Elias J. Rheingold, Joshua R. Williams, Benjamín J. Alemán, Christopher H. Hendon Direct electrochemical appraisal of black coffee quality using cyclic voltammetry In: Nature Communications, vol. 17, no. 3618, 2026, (Coffee flavor is primarily determined by the bean roast color and concentration of the beverage. Here, the authors show that both of these characteristics are reflected in the coffee’s cyclic voltammogram. This approach enables rapid determination of the strength and roast intensity of the coffee.). @article{nokey,
title = {Direct electrochemical appraisal of black coffee quality using cyclic voltammetry},
author = {Robin E. Bumbaugh and Doran L. Pennington and Lena C. Wehn and Elias J. Rheingold and Joshua R. Williams and Benjamín J. Alemán and Christopher H. Hendon },
url = {https://www.nature.com/articles/s41467-026-71526-5.pdf},
doi = {10.1038/s41467-026-71526-5},
year = {2026},
date = {2026-04-28},
journal = {Nature Communications},
volume = {17},
number = {3618},
abstract = {Despite coffee’s popularity, there are no quantitative methods to measure a chemical property of a black coffee drink in situ and relate it to a flavor experience. Here we show that cyclic voltammetry can be used without any additional sample preparation to directly measure the strength of a coffee beverage and, separately, how dark the coffee has been roasted; these two properties are implicated in the sensory profile of the beverage. We show that the current passed for the cathodic features that precede hydrogen evolution are linearly related to beverage strength. The same features are suppressed with subsequent cycling due to coffee material accumulating on the electrode. The magnitude of suppression is directly related to roast color, which dictates the ensemble chemical composition and flavor of the beverage. Together, this voltammetric analysis decouples beverage strength from roast color and offers a strategy for rapidly assessing flavor-correlated chemical properties of coffee.},
note = {Coffee flavor is primarily determined by the bean roast color and concentration of the beverage. Here, the authors show that both of these characteristics are reflected in the coffee’s cyclic voltammogram. This approach enables rapid determination of the strength and roast intensity of the coffee.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Despite coffee’s popularity, there are no quantitative methods to measure a chemical property of a black coffee drink in situ and relate it to a flavor experience. Here we show that cyclic voltammetry can be used without any additional sample preparation to directly measure the strength of a coffee beverage and, separately, how dark the coffee has been roasted; these two properties are implicated in the sensory profile of the beverage. We show that the current passed for the cathodic features that precede hydrogen evolution are linearly related to beverage strength. The same features are suppressed with subsequent cycling due to coffee material accumulating on the electrode. The magnitude of suppression is directly related to roast color, which dictates the ensemble chemical composition and flavor of the beverage. Together, this voltammetric analysis decouples beverage strength from roast color and offers a strategy for rapidly assessing flavor-correlated chemical properties of coffee. |
| 304. | | Sung Kwan Tae, Gabriella Pasya Irianti, Ye Chan Kim, Seong-Gyun Im, S. Joon Kwon, Su-Mi Hur, So Youn Kim Quantitative control of orientational and positional disorder in nanopatterned arrays of metal-infiltrated block copolymers In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Quantitative control of orientational and positional disorder in nanopatterned arrays of metal-infiltrated block copolymers},
author = {Sung Kwan Tae and Gabriella Pasya Irianti and Ye Chan Kim and Seong-Gyun Im and S. Joon Kwon and Su-Mi Hur and So Youn Kim },
url = {https://www.nature.com/articles/s41467-026-72360-5_reference.pdf},
doi = {10.1038/s41467-026-72360-5},
year = {2026},
date = {2026-04-28},
journal = {Nature Communications},
volume = {17},
abstract = {Correlated disorder is not uncommon in nature and often possesses unexpectedly unique properties, inspiring scientists to explore disorder as a functional design element. However, the controlled realization and reproduction of disordered nanostructures remain experimentally challenging, with the concept of “disorder” itself implying its multifaceted nature. Here, we present a methodology to tune structural disorder using metal-infiltrated block copolymers. Starting from a single-grain hexagonal lattice formed by sphere-forming block copolymer thin films, we intentionally introduce and modulate disorder by controlling annealing temperatures and the type of incorporated metals. We establish a robust analytical framework to quantify the order/disorder parameters, providing a clear yet precise assessment of structural irregularity. Supported by molecular dynamics simulations that reveal the mechanisms of disorder formation, we demonstrate a comprehensive dispersion spectrum of nanoparticles—ranging from highly ordered to disordered states. Supported by phononic bandgap calculations, we show that this continuum can serve as a platform for the controlled engineering of disordered wave-manipulating systems.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Correlated disorder is not uncommon in nature and often possesses unexpectedly unique properties, inspiring scientists to explore disorder as a functional design element. However, the controlled realization and reproduction of disordered nanostructures remain experimentally challenging, with the concept of “disorder” itself implying its multifaceted nature. Here, we present a methodology to tune structural disorder using metal-infiltrated block copolymers. Starting from a single-grain hexagonal lattice formed by sphere-forming block copolymer thin films, we intentionally introduce and modulate disorder by controlling annealing temperatures and the type of incorporated metals. We establish a robust analytical framework to quantify the order/disorder parameters, providing a clear yet precise assessment of structural irregularity. Supported by molecular dynamics simulations that reveal the mechanisms of disorder formation, we demonstrate a comprehensive dispersion spectrum of nanoparticles—ranging from highly ordered to disordered states. Supported by phononic bandgap calculations, we show that this continuum can serve as a platform for the controlled engineering of disordered wave-manipulating systems. |
| 303. | | Dominic J. P. Koyroytsaltis-McQuire, Shailendra K. Chaubey, Rahul Kumar, Paula L. Lalaguna, Tamas Javorfi, Giuliano Siligardi, Affar Karimullah, Adrian J. Lapthorn, Yoshito Y. Tanaka, Shun Hashiyada, Nikolaj Gadegaard, Malcolm Kadodwala Metasurface analogues of molecular diastereomers from hierarchical multiscale chiral interactions with biomolecules In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Metasurface analogues of molecular diastereomers from hierarchical multiscale chiral interactions with biomolecules},
author = {Dominic J. P. Koyroytsaltis-McQuire and Shailendra K. Chaubey and Rahul Kumar and Paula L. Lalaguna and Tamas Javorfi and Giuliano Siligardi and Affar Karimullah and Adrian J. Lapthorn and Yoshito Y. Tanaka and Shun Hashiyada and Nikolaj Gadegaard and Malcolm Kadodwala },
url = {https://www.nature.com/articles/s41467-026-72200-6_reference.pdf},
doi = {10.1038/s41467-026-72200-6},
year = {2026},
date = {2026-04-22},
urldate = {2026-04-22},
journal = {Nature Communications},
volume = {17},
abstract = {Chirality occurs at molecular and nanoscales, but these forms of handedness are usually treated independently. We demonstrate a hybrid chiral state, termed a meta-diastereomer, in which molecular chirality and nanoscale structural chirality are coupled at an interface to determine the optical response, with distinct states defined by the relative handedness of the two components, as in molecular diastereomers. Unlike strategies that rely on enhanced near-field optical chirality, the coupling here arises from a charged, polarisable interfacial layer that perturbs electromagnetic boundary conditions. The resulting reduction in symmetry renders linear optical observables stereostructurally informative, in the same way that molecular diastereomers can be distinguished using non-chiral measurement techniques. This behaviour distinguishes native versus denatured protein layers and differentiates protein-protein binding states in a model antigen-antibody system. These results identify electromagnetic boundary conditions as a mechanism by which chirality can be transmitted across length scales and provide a boundary-condition-based framework for hierarchical chirality at biomolecule-metasurface interfaces.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chirality occurs at molecular and nanoscales, but these forms of handedness are usually treated independently. We demonstrate a hybrid chiral state, termed a meta-diastereomer, in which molecular chirality and nanoscale structural chirality are coupled at an interface to determine the optical response, with distinct states defined by the relative handedness of the two components, as in molecular diastereomers. Unlike strategies that rely on enhanced near-field optical chirality, the coupling here arises from a charged, polarisable interfacial layer that perturbs electromagnetic boundary conditions. The resulting reduction in symmetry renders linear optical observables stereostructurally informative, in the same way that molecular diastereomers can be distinguished using non-chiral measurement techniques. This behaviour distinguishes native versus denatured protein layers and differentiates protein-protein binding states in a model antigen-antibody system. These results identify electromagnetic boundary conditions as a mechanism by which chirality can be transmitted across length scales and provide a boundary-condition-based framework for hierarchical chirality at biomolecule-metasurface interfaces. |
| 302. | | Xinhuan Zhang, Ziyang Wang, Ping Duan, Xijuan Wang, Yiming Chen, Yuxiao Chen, Manqiu Ma, Bailin Gao, Chuanxiang Chen, Zhichao Pan, Chuancheng Jia, Saisai Yuan Performance improvement of single-molecule sensors through deep learning-based decoding of tunneling signals enables sub-attomolar sensitivity In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Performance improvement of single-molecule sensors through deep learning-based decoding of tunneling signals enables sub-attomolar sensitivity},
author = {Xinhuan Zhang and Ziyang Wang and Ping Duan and Xijuan Wang and Yiming Chen and Yuxiao Chen and Manqiu Ma and Bailin Gao and Chuanxiang Chen and Zhichao Pan and Chuancheng Jia and Saisai Yuan },
url = {https://www.nature.com/articles/s41467-026-72249-3_reference.pdf},
doi = {10.1038/s41467-026-72249-3},
year = {2026},
date = {2026-04-21},
urldate = {2026-04-21},
journal = {Nature Communications},
volume = {17},
abstract = {Detecting individual molecules in real time provides high sensitivity for sensing applications. The break junction technique enables highly sensitive single-molecule detection by capturing the specific electronic signatures of individual molecules. However, this method is typically restricted by the requirement for anchoring groups on target analytes. Harnessing intermolecular interactions offers a solution to detect molecules without anchoring groups. Yet the resulting signals are often weak, sparse, and hidden in ensemble analyses. Here, we integrate rationally designed porphyrin-based probes with a time-frequency deep-learning framework to decode these subtle signatures. With this approach, we elevate the detection limit to the sub-attomolar level (10–18 mol L–1) with seconds-scale response time (26 s). Validated across diverse analytes, our strategy consistently enhances sensitivity, confirming its generalizability. This synergistic strategy establishes a paradigm for single-molecule chemical sensing both in methodological and performance dimensions. By transforming fleeting interactions into actionable detection, the approach could provide a robust and broadly applicable framework for environmental monitoring and molecular diagnostic.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Detecting individual molecules in real time provides high sensitivity for sensing applications. The break junction technique enables highly sensitive single-molecule detection by capturing the specific electronic signatures of individual molecules. However, this method is typically restricted by the requirement for anchoring groups on target analytes. Harnessing intermolecular interactions offers a solution to detect molecules without anchoring groups. Yet the resulting signals are often weak, sparse, and hidden in ensemble analyses. Here, we integrate rationally designed porphyrin-based probes with a time-frequency deep-learning framework to decode these subtle signatures. With this approach, we elevate the detection limit to the sub-attomolar level (10–18 mol L–1) with seconds-scale response time (26 s). Validated across diverse analytes, our strategy consistently enhances sensitivity, confirming its generalizability. This synergistic strategy establishes a paradigm for single-molecule chemical sensing both in methodological and performance dimensions. By transforming fleeting interactions into actionable detection, the approach could provide a robust and broadly applicable framework for environmental monitoring and molecular diagnostic. |
| 301. | | Sehee Yang, Sanghyuk Park, Yongseok Jung, Young Geon Kim, Jiwoo Lee, Shin-Hyun Kim Fractional crystallization of colloids by sequential tuning of depletion forces In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Fractional crystallization of colloids by sequential tuning of depletion forces},
author = {Sehee Yang and Sanghyuk Park and Yongseok Jung and Young Geon Kim and Jiwoo Lee and Shin-Hyun Kim },
url = {https://www.nature.com/articles/s41467-026-72000-y_reference.pdf},
doi = {10.1038/s41467-026-72000-y},
year = {2026},
date = {2026-04-15},
journal = {Nature Communications},
volume = {17},
abstract = {Fractional crystallization is a powerful and precise separation method capable of discriminating even between molecular isomers by exploiting subtle solubility differences. While colloidal particles are inherently insoluble, they undergo crystallization and melting transitions governed by interparticle interactions, akin to molecular systems. Inspired by this molecular approach, here we show a simple thermodynamic method to separate colloidal particles by size using depletion forces. By tuning interparticle potentials to emulate Mie-type interactions, we show that particles crystallize independently when the depth of the attractive well exceeds a species-specific threshold. Due to greater excluded volume overlap, larger particles experience stronger depletion attractions, allowing them to crystallize while smaller particles stay in the fluid. Building upon this principle, we establish a sequential fractional crystallization protocol—comprising crystallization, melting, and recrystallization—that achieves fractionation of colloids with <10% size differences. Notably, when all particle species marginally exceed the crystallization threshold, they preferentially crystallize into their respective lattices to maximize coordination numbers and enthalpic gains. Under stronger attractions, particles first form kinetically arrested glassy alloys that later relax into metastable crystalline alloys without macroscopic separation.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fractional crystallization is a powerful and precise separation method capable of discriminating even between molecular isomers by exploiting subtle solubility differences. While colloidal particles are inherently insoluble, they undergo crystallization and melting transitions governed by interparticle interactions, akin to molecular systems. Inspired by this molecular approach, here we show a simple thermodynamic method to separate colloidal particles by size using depletion forces. By tuning interparticle potentials to emulate Mie-type interactions, we show that particles crystallize independently when the depth of the attractive well exceeds a species-specific threshold. Due to greater excluded volume overlap, larger particles experience stronger depletion attractions, allowing them to crystallize while smaller particles stay in the fluid. Building upon this principle, we establish a sequential fractional crystallization protocol—comprising crystallization, melting, and recrystallization—that achieves fractionation of colloids with <10% size differences. Notably, when all particle species marginally exceed the crystallization threshold, they preferentially crystallize into their respective lattices to maximize coordination numbers and enthalpic gains. Under stronger attractions, particles first form kinetically arrested glassy alloys that later relax into metastable crystalline alloys without macroscopic separation. |
| 300. | | Byeong-Gyu Chae, Mihye Lim, Junho Lee, Nayoun Won, Soo Kyung Kwon, Ara Jo, Dong Jin Yun, Sangjun Lee, Jwa-Min Nam, Soohwan Sul, Tae-Gon Kim 3D mapping of compositional gradients of core-shell structures in AgInxGa1-xS2 quantum dots by atom probe tomography In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {3D mapping of compositional gradients of core-shell structures in AgInxGa1-xS2 quantum dots by atom probe tomography},
author = {Byeong-Gyu Chae and Mihye Lim and Junho Lee and Nayoun Won and Soo Kyung Kwon and Ara Jo and Dong Jin Yun and Sangjun Lee and Jwa-Min Nam and Soohwan Sul and Tae-Gon Kim },
url = {https://www.nature.com/articles/s41467-026-71518-5_reference.pdf},
doi = {10.1038/s41467-026-71518-5},
year = {2026},
date = {2026-04-03},
journal = {Nature Communications},
volume = {17},
abstract = {Colloidal quantum dots (QDs), which exhibit tunable band gaps depending on their size and composition, are widely studied for light-emitting and optoelectronic applications. AgInxGa1-xS2-based QDs are particularly promising due to their pure green emission, high blue absorption, and environmental friendliness. However, a comprehensive understanding of these quaternary QDs remains challenging because of the difficulty in examining their complex compositional structure. Here, we three-dimensionally characterize quaternary QDs (AgInxGa1-xS2-based heterostructured QDs with core/shell and core/shell/shell structures) on the atomic scale using atom probe tomography. We reveal that both the AgInxGa1-xS2/AgGaS2 QDs with and without an outer ZnS shell have compositional gradients at their interfaces and elemental inhomogeneity among their cores. Furthermore, an Ag-deficient AgInyGa1-yS2 layer is identified on the outer surface of the AgGaS2 shell, where the stoichiometric fractions satisfy x » y, arising from differences in the precursor reactivity. Meanwhile, in the AgInxGa1-xS2/AgGaS2/ZnS QDs, the outer ZnS shell evolves into Zn1-3/2xGaxS through a cation exchange process, ensuring structural and chemical compatibility with the inner shell. Our findings uncover the internal architecture and nanoscale elemental distributions of quaternary QDs, providing guidance for the future development of QDs.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Colloidal quantum dots (QDs), which exhibit tunable band gaps depending on their size and composition, are widely studied for light-emitting and optoelectronic applications. AgInxGa1-xS2-based QDs are particularly promising due to their pure green emission, high blue absorption, and environmental friendliness. However, a comprehensive understanding of these quaternary QDs remains challenging because of the difficulty in examining their complex compositional structure. Here, we three-dimensionally characterize quaternary QDs (AgInxGa1-xS2-based heterostructured QDs with core/shell and core/shell/shell structures) on the atomic scale using atom probe tomography. We reveal that both the AgInxGa1-xS2/AgGaS2 QDs with and without an outer ZnS shell have compositional gradients at their interfaces and elemental inhomogeneity among their cores. Furthermore, an Ag-deficient AgInyGa1-yS2 layer is identified on the outer surface of the AgGaS2 shell, where the stoichiometric fractions satisfy x » y, arising from differences in the precursor reactivity. Meanwhile, in the AgInxGa1-xS2/AgGaS2/ZnS QDs, the outer ZnS shell evolves into Zn1-3/2xGaxS through a cation exchange process, ensuring structural and chemical compatibility with the inner shell. Our findings uncover the internal architecture and nanoscale elemental distributions of quaternary QDs, providing guidance for the future development of QDs. |
| 299. | | Faisal Abbas, Mohammad Salehian, Peter Hou, Jonathan Moores, Jonathan Goldie, Alexandros Tsioutsios, Theo Tait, Victor Portela, Quentin Boulay, Roland Thiolliere, Ashley Stark, Jean-Jacques Schwartz, Jerome Guerin, Andrew G. P. Maloney, Alexandru A. Moldovan, Gavin K. Reynolds, Jérôme Mantanus, Catriona Clark, Paul Chapman, Alastair Florence, Daniel Markl Accelerated drug development using a digital formulator and a self-driving tableting data factory In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Accelerated drug development using a digital formulator and a self-driving tableting data factory},
author = {Faisal Abbas and Mohammad Salehian and Peter Hou and Jonathan Moores and Jonathan Goldie and Alexandros Tsioutsios and Theo Tait and Victor Portela and Quentin Boulay and Roland Thiolliere and Ashley Stark and Jean-Jacques Schwartz and Jerome Guerin and Andrew G. P. Maloney and Alexandru A. Moldovan and Gavin K. Reynolds and Jérôme Mantanus and Catriona Clark and Paul Chapman and Alastair Florence and Daniel Markl },
url = {https://www.nature.com/articles/s41467-026-71204-6_reference.pdf},
doi = {10.1038/s41467-026-71204-6},
year = {2026},
date = {2026-04-01},
journal = {Nature Communications},
volume = {17},
abstract = {Advances in drug discovery and clinical research have shifted the bottleneck in medicines development to chemistry, manufacturing, and controls activities, a critically step for regulatory approval. This includes formulation and process development of a new drug product, which traditionally requires extensive resources, often leading to suboptimal outcomes. These development processes must adapt to follow the advances in drug discovery and clinical research and ultimately shorten timelines while ensuring product quality and safety. In this work, we present an integrated platform for tablet formulation and process development that couples a digital formulator, an in-silico optimisation tool using a predictive material-to-tablet model, with a self-driving tableting data factory, which applies Bayesian optimisation within an automated, fully integrated per-tablet manufacturing to testing workflow. The results demonstrate a reduction in the time from material characterisation to in-specification tablets to 6 h and a reduction in API material use by 65% compared to current state-of-the-art methods.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Advances in drug discovery and clinical research have shifted the bottleneck in medicines development to chemistry, manufacturing, and controls activities, a critically step for regulatory approval. This includes formulation and process development of a new drug product, which traditionally requires extensive resources, often leading to suboptimal outcomes. These development processes must adapt to follow the advances in drug discovery and clinical research and ultimately shorten timelines while ensuring product quality and safety. In this work, we present an integrated platform for tablet formulation and process development that couples a digital formulator, an in-silico optimisation tool using a predictive material-to-tablet model, with a self-driving tableting data factory, which applies Bayesian optimisation within an automated, fully integrated per-tablet manufacturing to testing workflow. The results demonstrate a reduction in the time from material characterisation to in-specification tablets to 6 h and a reduction in API material use by 65% compared to current state-of-the-art methods. |
| 298. | | Mingrui Zhou, Wen Wang, Jinyi Sun, Yuze Yu, Meng Nie, Hubiao Huang, Haimei Zheng, Tao Xu, Litao Sun Accelerated directional growth of seaweed-like iron oxide branches driven by localized electric fields of gold nanoparticles in liquid In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Accelerated directional growth of seaweed-like iron oxide branches driven by localized electric fields of gold nanoparticles in liquid},
author = {Mingrui Zhou and Wen Wang and Jinyi Sun and Yuze Yu and Meng Nie and Hubiao Huang and Haimei Zheng and Tao Xu and Litao Sun },
url = {https://www.nature.com/articles/s41467-026-71352-9_reference.pdf},
doi = {10.1038/s41467-026-71352-9},
year = {2026},
date = {2026-03-31},
journal = {Nature Communications},
volume = {17},
abstract = {Branched nanostructures have attracted significant attention due to their potential applications across diverse fields. Precise control over branched morphology is essential for enhancing their functionality, yet it remains a considerable challenge. In this work, in-situ liquid-cell transmission electron microscopy (LCTEM) is employed to investigate the controllable growth of seaweed-like iron oxide branches in the presence of charged gold nanoparticles (Au NPs) within an organic solution. In contrast to the conventional tip-splitting behavior observed in the absence of Au NPs, the branches exhibit directional and accelerated growth toward the Au NPs without further splitting. Finite-element analysis reveals that the local electric field between the charged Au NPs and the branches promotes reactant aggregation at the branch tips, thereby driving their directional and accelerated growth. This study provides insights into the growth mechanisms of seaweed-like nanostructures and highlights the potential of local electric fields for morphological control of branched structures.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Branched nanostructures have attracted significant attention due to their potential applications across diverse fields. Precise control over branched morphology is essential for enhancing their functionality, yet it remains a considerable challenge. In this work, in-situ liquid-cell transmission electron microscopy (LCTEM) is employed to investigate the controllable growth of seaweed-like iron oxide branches in the presence of charged gold nanoparticles (Au NPs) within an organic solution. In contrast to the conventional tip-splitting behavior observed in the absence of Au NPs, the branches exhibit directional and accelerated growth toward the Au NPs without further splitting. Finite-element analysis reveals that the local electric field between the charged Au NPs and the branches promotes reactant aggregation at the branch tips, thereby driving their directional and accelerated growth. This study provides insights into the growth mechanisms of seaweed-like nanostructures and highlights the potential of local electric fields for morphological control of branched structures. |
| 297. | | Shuang Li, Zhongliang Cao, Haolong Chang, Wei Zheng, Guoao Luo, Lifeng Zhang, Weiwei Zhang, Langli Luo, Xing Chen Copper-hydroxyl interactions drive water-promoted copper surface oxidation and mobility In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Copper-hydroxyl interactions drive water-promoted copper surface oxidation and mobility},
author = {Shuang Li and Zhongliang Cao and Haolong Chang and Wei Zheng and Guoao Luo and Lifeng Zhang and Weiwei Zhang and Langli Luo and Xing Chen },
url = {https://www.nature.com/articles/s41467-026-70947-6_reference.pdf},
doi = {10.1038/s41467-026-70947-6},
year = {2026},
date = {2026-03-24},
journal = {Nature Communications},
volume = {17},
abstract = {The reactions of oxygen (O2) and water (H2O) molecules with metal surfaces are critical to heterogeneous catalysis, corrosion, and electrochemical energy conversion. However, disentangling their individual roles remains challenging because both pathways yield the same dissociation product, atomic oxygen (O), and share the hydroxyl (OH) intermediate, thereby obscuring the molecular origin of metal oxidation. In this study, we combine in-situ transmission electron microscopy techniques and ReaxFF reactive force field molecular dynamics (MD) simulations to elucidate the promotional role of H2O in copper (Cu) surface oxidation. Our results reveal that the structurally disordered Cu/CuOx interface preferentially adsorbs OH derived from H2O dissociation. The resulting strong Cu-OH interaction causes dynamic disorder in the topmost Cu layer while enriching electron density in the sublayer. This coupled structural and electronic modulation lowers the resistance for oxygen incorporation, promoting deeper lattice penetration, accelerating oxidation, and enhancing Cu atomic mobility. In contrast, oxidation under pure O2 produces a comparatively ordered interface that suppresses sustained oxygen ingress, rendering further oxidation kinetically less favorable. These findings identify OH-mediated interfacial dynamics as a key driver of water-assisted metal oxidation and provide mechanistic guidance for controlling oxidation processes in catalytic and corrosion-resistant materials.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The reactions of oxygen (O2) and water (H2O) molecules with metal surfaces are critical to heterogeneous catalysis, corrosion, and electrochemical energy conversion. However, disentangling their individual roles remains challenging because both pathways yield the same dissociation product, atomic oxygen (O), and share the hydroxyl (OH) intermediate, thereby obscuring the molecular origin of metal oxidation. In this study, we combine in-situ transmission electron microscopy techniques and ReaxFF reactive force field molecular dynamics (MD) simulations to elucidate the promotional role of H2O in copper (Cu) surface oxidation. Our results reveal that the structurally disordered Cu/CuOx interface preferentially adsorbs OH derived from H2O dissociation. The resulting strong Cu-OH interaction causes dynamic disorder in the topmost Cu layer while enriching electron density in the sublayer. This coupled structural and electronic modulation lowers the resistance for oxygen incorporation, promoting deeper lattice penetration, accelerating oxidation, and enhancing Cu atomic mobility. In contrast, oxidation under pure O2 produces a comparatively ordered interface that suppresses sustained oxygen ingress, rendering further oxidation kinetically less favorable. These findings identify OH-mediated interfacial dynamics as a key driver of water-assisted metal oxidation and provide mechanistic guidance for controlling oxidation processes in catalytic and corrosion-resistant materials. |
| 296. | | Sebastian Hartweg, Dušan K. Božanić, Gustavo A. Garcia, Laurent Nahon Linking photoelectron circular dichroism to the asymmetric total photoemission yield measured in aerosol nanoparticles of tyrosine In: Nature Communications, vol. 17, no. 2792, 2026, (Photoelectron circular dichroism (PECD) is a strong chiroptical effect affecting the angular distribution of photoelectrons emitted from chiral molecules ionized by circularly polarized light. Here, the authors show how PECD creates a similar-magnitude effect in the total photoemission yield of condensed particles.). @article{nokey,
title = {Linking photoelectron circular dichroism to the asymmetric total photoemission yield measured in aerosol nanoparticles of tyrosine},
author = {Sebastian Hartweg and Dušan K. Božanić and Gustavo A. Garcia and Laurent Nahon },
url = {https://www.nature.com/articles/s41467-026-70997-w.pdf},
doi = {10.1038/s41467-026-70997-w},
year = {2026},
date = {2026-03-24},
urldate = {2026-03-24},
journal = {Nature Communications},
volume = {17},
number = {2792},
abstract = {Spectroscopic techniques that are sensitive to molecular chirality are important analytical tools to quantitatively determine enantiomeric excess and purity of chiral molecular samples. Many chiroptical processes however produce weak enantio-specific asymmetries due to their origin relying on weak magnetic dipole or electric quadrupole effects. Photoelectron circular dichroism (PECD) in contrast, is an intense effect, that is fully contained in the electric dipole description of light matter interaction and creates a chiral asymmetry in the photoelectron angular distribution. Here, we demonstrate that this chiral signature in the angular distribution of emitted electrons can be translated into the total photoemission yield for particulate matter. The resulting chiral asymmetry of the photoemission yield (CAPY), mediated by the attenuation of light within condensed particles, can be detected experimentally without requiring high vacuum systems and electron spectrometers. This effect can be exploited as an analytical tool with high sensitivity to chirality and enantiopurity for studies of chiral organic and hybrid submicron particles in environmental, biomedical or catalytic applications.},
note = {Photoelectron circular dichroism (PECD) is a strong chiroptical effect affecting the angular distribution of photoelectrons emitted from chiral molecules ionized by circularly polarized light. Here, the authors show how PECD creates a similar-magnitude effect in the total photoemission yield of condensed particles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Spectroscopic techniques that are sensitive to molecular chirality are important analytical tools to quantitatively determine enantiomeric excess and purity of chiral molecular samples. Many chiroptical processes however produce weak enantio-specific asymmetries due to their origin relying on weak magnetic dipole or electric quadrupole effects. Photoelectron circular dichroism (PECD) in contrast, is an intense effect, that is fully contained in the electric dipole description of light matter interaction and creates a chiral asymmetry in the photoelectron angular distribution. Here, we demonstrate that this chiral signature in the angular distribution of emitted electrons can be translated into the total photoemission yield for particulate matter. The resulting chiral asymmetry of the photoemission yield (CAPY), mediated by the attenuation of light within condensed particles, can be detected experimentally without requiring high vacuum systems and electron spectrometers. This effect can be exploited as an analytical tool with high sensitivity to chirality and enantiopurity for studies of chiral organic and hybrid submicron particles in environmental, biomedical or catalytic applications. |
| 295. | | Hyeokjun Kim, Hyobin Ham, Chang Hyeok Lim, Jin Su Park, SeungHwan Roh, Hak June Lee, Myeongjae Lee, Jinho Keum, Se Young Park, Jeong Woo Park, Seongjae Lee, Hajin Bhang, Seunghan Lee, Hyunwoo Jo, Yong Hyun Jo, Jin-Wook Shin, Wan Ki Bae, Chan-mo Kang, Moon Sung Kang, BongSoo Kim Photoresist-guided indirect photopatterning of quantum dots via carbene-mediated ligand thermocrosslinking In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Photoresist-guided indirect photopatterning of quantum dots via carbene-mediated ligand thermocrosslinking},
author = {Hyeokjun Kim and Hyobin Ham and Chang Hyeok Lim and Jin Su Park and SeungHwan Roh and Hak June Lee and Myeongjae Lee and Jinho Keum and Se Young Park and Jeong Woo Park and Seongjae Lee and Hajin Bhang and Seunghan Lee and Hyunwoo Jo and Yong Hyun Jo and Jin-Wook Shin and Wan Ki Bae and Chan-mo Kang and Moon Sung Kang and BongSoo Kim },
url = {https://www.nature.com/articles/s41467-026-70770-z_reference.pdf},
doi = {10.1038/s41467-026-70770-z},
year = {2026},
date = {2026-03-19},
journal = {Nature Communications},
volume = {17},
abstract = {Colloidal quantum dots (QDs) are leading candidates for next-generation optoelectronics owing to their tuneable bandgaps, narrow emission linewidths, and high luminescence quantum yields. For virtual-, augmented-, and mixed-reality display applications of these materials, patterning full-color QDs at μm-length scales is essential. However, existing photolithographic approaches often degrade QD luminance characteristics by exposing them to harsh processing conditions, or they compromise the structural fidelity of the resulting patterns. Here we report a photoresist-guided indirect (PIN) photopatterning strategy that includes (i) lithographic formation of sacrificial PR patterns, (ii) deposition of a crosslinked QD film on top, and (iii) PR stripping that removes the sacrificial PR, leaving behind crosslinked QD patterns on the substrate. QD crosslinking is mediated by a diazo-based ligand thermocrosslinker, Diazo-4-LiXer. Leveraging low-temperature (110–120 °C)-activated carbene chemistry, Diazo-4-LiXer bridges neighbouring QDs while maintaining their intrinsic photoluminescence and electroluminescence through repeated processing. Moreover, Diazo-4-LiXer enables thermocrosslinking without affecting the underlying photoresist pre-patterns, which serve as structural templates determining the thickness and fidelity of the QD patterns. Using PIN photopatterning, we realize high-fidelity RGB patterns exceeding 4,000 pixels per inch resolution and demonstrate integration-level scalability by fabricating a 10 × 10 passive-matrix full-colour RGB QD–LED array.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Colloidal quantum dots (QDs) are leading candidates for next-generation optoelectronics owing to their tuneable bandgaps, narrow emission linewidths, and high luminescence quantum yields. For virtual-, augmented-, and mixed-reality display applications of these materials, patterning full-color QDs at μm-length scales is essential. However, existing photolithographic approaches often degrade QD luminance characteristics by exposing them to harsh processing conditions, or they compromise the structural fidelity of the resulting patterns. Here we report a photoresist-guided indirect (PIN) photopatterning strategy that includes (i) lithographic formation of sacrificial PR patterns, (ii) deposition of a crosslinked QD film on top, and (iii) PR stripping that removes the sacrificial PR, leaving behind crosslinked QD patterns on the substrate. QD crosslinking is mediated by a diazo-based ligand thermocrosslinker, Diazo-4-LiXer. Leveraging low-temperature (110–120 °C)-activated carbene chemistry, Diazo-4-LiXer bridges neighbouring QDs while maintaining their intrinsic photoluminescence and electroluminescence through repeated processing. Moreover, Diazo-4-LiXer enables thermocrosslinking without affecting the underlying photoresist pre-patterns, which serve as structural templates determining the thickness and fidelity of the QD patterns. Using PIN photopatterning, we realize high-fidelity RGB patterns exceeding 4,000 pixels per inch resolution and demonstrate integration-level scalability by fabricating a 10 × 10 passive-matrix full-colour RGB QD–LED array. |
| 294. | | Mengya Zhang, Tongcheng Yu, Hao Liu, Chao Lin, Yaping Yang, Bowen Lv, Qi Zhang, Ming Chen, Tianshuai Wang, Weihong Hua, Kai Han Heat-assisted hot-hole transfer increases the surface-enhanced Raman activity of Au-TiO2 nanoarrays In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Heat-assisted hot-hole transfer increases the surface-enhanced Raman activity of Au-TiO2 nanoarrays},
author = {Mengya Zhang and Tongcheng Yu and Hao Liu and Chao Lin and Yaping Yang and Bowen Lv and Qi Zhang and Ming Chen and Tianshuai Wang and Weihong Hua and Kai Han },
url = {https://www.nature.com/articles/s41467-026-70822-4_reference.pdf},
doi = {10.1038/s41467-026-70822-4},
year = {2026},
date = {2026-03-17},
journal = {Nature Communications},
volume = {17},
abstract = {Monitoring the evolution of molecules during photo and thermal synergistically induced physical and chemical processes is of paramount interest in fields including chemical, material, and energy research. Surface-enhanced Raman spectroscopy (SERS) is a highly promising technology in this regard, offering advantages of sensitivity, real-time, and label-free detection. However, the application of conventional SERS in high-temperature environments has faced challenges due to the inevitable loss of activity and decline in sensitivity. Herein, we synthesize Au-TiO2 nanoarrays as SERS substrates, and an anomalous enhancement of Raman signal with increasing temperature is observed. The signal intensity increases by 11.41 times at 180 °C compared to that at 22 °C. This high-temperature enhancement in Raman activity is attributed to an underlying mechanism: heat-assisted hot-hole transfer, which enables 785 nm photon-induced hot-hole transfer from Au to TiO2. Our work expands the application of the SERS technique for high-temperature chemical analysis and molecular diagnostics.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Monitoring the evolution of molecules during photo and thermal synergistically induced physical and chemical processes is of paramount interest in fields including chemical, material, and energy research. Surface-enhanced Raman spectroscopy (SERS) is a highly promising technology in this regard, offering advantages of sensitivity, real-time, and label-free detection. However, the application of conventional SERS in high-temperature environments has faced challenges due to the inevitable loss of activity and decline in sensitivity. Herein, we synthesize Au-TiO2 nanoarrays as SERS substrates, and an anomalous enhancement of Raman signal with increasing temperature is observed. The signal intensity increases by 11.41 times at 180 °C compared to that at 22 °C. This high-temperature enhancement in Raman activity is attributed to an underlying mechanism: heat-assisted hot-hole transfer, which enables 785 nm photon-induced hot-hole transfer from Au to TiO2. Our work expands the application of the SERS technique for high-temperature chemical analysis and molecular diagnostics. |
| 293. | | Sakshi Yadav Schmid, Benjamin Helfrecht, Amy Stegmann, Benjamin A. Legg, Harley Pyles, Jiajun Chen, John R. Edison, Maxim Ziatdinov, Zdenek Preisler, Orion Dollar, Stephen Whitelam, Sergei Kalinin, David Baker, Christopher J. Mundy, Shuai Zhang, James J. De Yoreo Impact of solvent forces and broken symmetry on the assembly of designed proteins at a liquid-solid interface In: Nature Communications, vol. 17, no. 2446, 2026, (Manipulating chemical interactions is crucial in protein assembly. Here, the authors highlight the importance of accounting for solvent forces mediated by interfacial hydration structures when designing protein assemblies at liquid-crystal interfaces.). @article{nokey,
title = {Impact of solvent forces and broken symmetry on the assembly of designed proteins at a liquid-solid interface},
author = {Sakshi Yadav Schmid and Benjamin Helfrecht and Amy Stegmann and Benjamin A. Legg and Harley Pyles and Jiajun Chen and John R. Edison and Maxim Ziatdinov and Zdenek Preisler and Orion Dollar and Stephen Whitelam and Sergei Kalinin and David Baker and Christopher J. Mundy and Shuai Zhang and James J. De Yoreo },
url = {https://www.nature.com/articles/s41467-026-69170-0.pdf},
doi = {10.1038/s41467-026-69170-0},
year = {2026},
date = {2026-03-13},
journal = {Nature Communications},
volume = {17},
number = {2446},
abstract = {The era of protein design has enabled the creation of hybrid protein-inorganic interfaces, leading to both surface-directed self-assembly of de novo protein architectures and protein-directed formation of inorganic materials. However, the resulting patterns of protein assembly are often unexpected, implying that essential interactions are not accounted for in current design platforms. Here, we use high-speed atomic force microscopy (AFM) analyzed through machine learning to follow the assembly of protein nanorods in aqueous electrolytes on two types of mica exhibiting disparate symmetry elements, which are imprinted on the overlying hydration structure. Using Monte Carlo simulations, we reproduce the observed phases and show that an observed smectic phase, previously thought to be unstable for non-interacting rods in two dimensions, emerges when crystal symmetry introduces a directional bias. The findings demonstrate the importance of incorporating solvent forces as modulated by the hydration structure inherent to interfacial systems when designing protein assemblies at liquid-crystal interfaces. Coupling physics-based simulations that can account for these factors to de novo protein design algorithms can lead to improved design platforms for bio-inspired, hybrid materials.},
note = {Manipulating chemical interactions is crucial in protein assembly. Here, the authors highlight the importance of accounting for solvent forces mediated by interfacial hydration structures when designing protein assemblies at liquid-crystal interfaces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The era of protein design has enabled the creation of hybrid protein-inorganic interfaces, leading to both surface-directed self-assembly of de novo protein architectures and protein-directed formation of inorganic materials. However, the resulting patterns of protein assembly are often unexpected, implying that essential interactions are not accounted for in current design platforms. Here, we use high-speed atomic force microscopy (AFM) analyzed through machine learning to follow the assembly of protein nanorods in aqueous electrolytes on two types of mica exhibiting disparate symmetry elements, which are imprinted on the overlying hydration structure. Using Monte Carlo simulations, we reproduce the observed phases and show that an observed smectic phase, previously thought to be unstable for non-interacting rods in two dimensions, emerges when crystal symmetry introduces a directional bias. The findings demonstrate the importance of incorporating solvent forces as modulated by the hydration structure inherent to interfacial systems when designing protein assemblies at liquid-crystal interfaces. Coupling physics-based simulations that can account for these factors to de novo protein design algorithms can lead to improved design platforms for bio-inspired, hybrid materials. |
| 292. | | Jiada Fan, Zhuang Ying, Jicun Ma, Jialiang Xu, Hui Cai, Jing Liu, Xinyue Wu, Chenhao Yang, Haorong Jiao, Qiulian Mao, Mei Chen, Liantuan Xiao, Yonggang Peng, Guofeng Zhang, Jiabin Cui, Mingyuan Gao Template-free synthesis of colloidal quantum dot assemblies with molecule-like architectures In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Template-free synthesis of colloidal quantum dot assemblies with molecule-like architectures},
author = {Jiada Fan and Zhuang Ying and Jicun Ma and Jialiang Xu and Hui Cai and Jing Liu and Xinyue Wu and Chenhao Yang and Haorong Jiao and Qiulian Mao and Mei Chen and Liantuan Xiao and Yonggang Peng and Guofeng Zhang and Jiabin Cui and Mingyuan Gao },
url = {https://www.nature.com/articles/s41467-026-70555-4_reference.pdf},
doi = {10.1038/s41467-026-70555-4},
year = {2026},
date = {2026-03-13},
urldate = {2026-03-13},
journal = {Nature Communications},
volume = {17},
abstract = {Designing colloidal assemblies with molecule-like architectures comprising more than two electronically coupled quantum dots has often required technologically complex, expensive, and top-down nanofabrication. Here we demonstrate a one-pot chemical synthesis of dimeric, trimeric, and tetrameric assemblies of coupled molecule-like quantum dots (CMQDs), using ZnSe@ZnS quantum dots as model. We show that the “valence” of these “artificial atoms” can be readily tuned by the amount of a suitable ligand in the reaction mixture, and that high-temperature fusion yields highly ordered oriented attachment and strong electron coupling between bound QDs. The shapes of the fused assemblies echo the canonical sp-, sp²-, and sp³-hybridization motifs and can be interpreted as appropriately shaped confining potential wells for electrons and holes. This work establishes an experimentally accessible entry point to related “artificial molecules” with controllable chemical composition, geometry, and electronic structure. The enhanced or emergent properties of such nanomaterials are anticipated to advance applications in optoelectronics, sensing, and quantum photonic technologies.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Designing colloidal assemblies with molecule-like architectures comprising more than two electronically coupled quantum dots has often required technologically complex, expensive, and top-down nanofabrication. Here we demonstrate a one-pot chemical synthesis of dimeric, trimeric, and tetrameric assemblies of coupled molecule-like quantum dots (CMQDs), using ZnSe@ZnS quantum dots as model. We show that the “valence” of these “artificial atoms” can be readily tuned by the amount of a suitable ligand in the reaction mixture, and that high-temperature fusion yields highly ordered oriented attachment and strong electron coupling between bound QDs. The shapes of the fused assemblies echo the canonical sp-, sp²-, and sp³-hybridization motifs and can be interpreted as appropriately shaped confining potential wells for electrons and holes. This work establishes an experimentally accessible entry point to related “artificial molecules” with controllable chemical composition, geometry, and electronic structure. The enhanced or emergent properties of such nanomaterials are anticipated to advance applications in optoelectronics, sensing, and quantum photonic technologies. |
| 291. | | In Hwan Jung, Chetan C. Revadekar, Hag Sung Lee, Hyerim Hwang, Hyosung An, Bum Jun Park Nonequilibrium ordering dynamics of confined soft alginate hydrogel colloids driven by time-evolving electrostatic interactions In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Nonequilibrium ordering dynamics of confined soft alginate hydrogel colloids driven by time-evolving electrostatic interactions},
author = {In Hwan Jung and Chetan C. Revadekar and Hag Sung Lee and Hyerim Hwang and Hyosung An and Bum Jun Park },
url = {https://www.nature.com/articles/s41467-026-70266-w_reference.pdf},
doi = {10.1038/s41467-026-70266-w},
year = {2026},
date = {2026-03-09},
urldate = {2026-03-03},
journal = {Nature Communications},
volume = {17},
abstract = {Elucidating how repulsive interactions evolve to generate ordered structures in nonequilibrium colloidal systems remains a central challenge, partly because few experimental platforms provide particle-resolved access to their structural evolution. Here we show that alginate hydrogel colloids confined within cyclohexyl bromide (CHB) emulsion droplets form a controllable model system in which electrostatic interactions evolve in time and drive ordering. Ba2+ ions diffusing from the surrounding aqueous phase progressively crosslink the alginate droplets, increasing their surface charge, while buoyancy compacts them into locally quasi-two-dimensional layers within the CHB phase. As electrostatic repulsion strengthens, the assembly evolves from a disordered state to a hexagonally ordered structure. By calibrating Brownian dynamics simulations to the experimentally measured lattice spring constant, we constrain the effective Debye screening length to ≈2.5–3 μm. Quantitative imaging further shows that ordering emerges once a dimensionless interaction parameter—defined as the ratio of electrostatic interaction energy to thermal energy—reaches values of ≈117–149. The ordered state exhibits reversible disordered–order behavior under mechanical and magnetic perturbations, demonstrating a robust nonequilibrium platform for probing charge-regulated colloidal ordering under confinement.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Elucidating how repulsive interactions evolve to generate ordered structures in nonequilibrium colloidal systems remains a central challenge, partly because few experimental platforms provide particle-resolved access to their structural evolution. Here we show that alginate hydrogel colloids confined within cyclohexyl bromide (CHB) emulsion droplets form a controllable model system in which electrostatic interactions evolve in time and drive ordering. Ba2+ ions diffusing from the surrounding aqueous phase progressively crosslink the alginate droplets, increasing their surface charge, while buoyancy compacts them into locally quasi-two-dimensional layers within the CHB phase. As electrostatic repulsion strengthens, the assembly evolves from a disordered state to a hexagonally ordered structure. By calibrating Brownian dynamics simulations to the experimentally measured lattice spring constant, we constrain the effective Debye screening length to ≈2.5–3 μm. Quantitative imaging further shows that ordering emerges once a dimensionless interaction parameter—defined as the ratio of electrostatic interaction energy to thermal energy—reaches values of ≈117–149. The ordered state exhibits reversible disordered–order behavior under mechanical and magnetic perturbations, demonstrating a robust nonequilibrium platform for probing charge-regulated colloidal ordering under confinement. |
| 290. | | Yu Liu, Haoyang Hu, Yueheng Han, Jia Song Deon Chon, Chin Lee Lo, Zhixuan Chen, Zheng Zhang, Zhihong Yuan, Jun Ge
Accelerated discovery of highly active enzyme nanohybrids with parallelized Bayesian optimization in hybrid space In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Accelerated discovery of highly active enzyme nanohybrids with parallelized Bayesian optimization in hybrid space},
author = {Yu Liu and Haoyang Hu and Yueheng Han and Jia Song Deon Chon and Chin Lee Lo and Zhixuan Chen and Zheng Zhang and Zhihong Yuan and Jun Ge
},
url = {https://www.nature.com/articles/s41467-026-70251-3_reference.pdf},
doi = {10.1038/s41467-026-70251-3},
year = {2026},
date = {2026-03-07},
journal = {Nature Communications},
volume = {17},
abstract = {Artificial intelligence (AI) has significantly advanced protein engineering, enabling rapid enzyme evolution for diverse applications. However, the fragile nature of biomacromolecules requires enzyme immobilization to preserve catalytic activity under harsh industrial conditions, which often restricts substrate diffusion and reduces enzymatic activity. This challenge demands extensive trial-and-error experiments to optimize immobilized carriers with high activity for different enzymes. Here we show a machine-learning-guided workflow along with an algorithm named parallelized hybrid-space Bayesian optimization (PHBO) to accelerate the discovery of nanocarriers for specific enzymes and reactions. Leveraging prior knowledge, machine learning and iterative feedback, within limited number of experiments, this workflow explores the reaction space of over 107 experiments and achieves activity recovery of 100%, 90%, and 79% for glucose oxidase, catalase, and Candida Antarctica lipase B, respectively. These results demonstrate that data-efficient optimization can substantially accelerate the discovery of enzyme nanohybrids with high catalytic activity across diverse enzymatic systems.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Artificial intelligence (AI) has significantly advanced protein engineering, enabling rapid enzyme evolution for diverse applications. However, the fragile nature of biomacromolecules requires enzyme immobilization to preserve catalytic activity under harsh industrial conditions, which often restricts substrate diffusion and reduces enzymatic activity. This challenge demands extensive trial-and-error experiments to optimize immobilized carriers with high activity for different enzymes. Here we show a machine-learning-guided workflow along with an algorithm named parallelized hybrid-space Bayesian optimization (PHBO) to accelerate the discovery of nanocarriers for specific enzymes and reactions. Leveraging prior knowledge, machine learning and iterative feedback, within limited number of experiments, this workflow explores the reaction space of over 107 experiments and achieves activity recovery of 100%, 90%, and 79% for glucose oxidase, catalase, and Candida Antarctica lipase B, respectively. These results demonstrate that data-efficient optimization can substantially accelerate the discovery of enzyme nanohybrids with high catalytic activity across diverse enzymatic systems. |
| 289. | | Bingzheng Yan, D. Sulalith N. D. Samarasinghe, Jing Sun, Hongwen Deng, Lei Li, Ming-Qiang Qi, Fangming Zhao, Qinghua Xu, Huifang Guo, Xueli Sun, Xuekun Gong, Rong Huo, Mengsi Zhu, Qingyuan Wu, Zhenlang Xie, Chengrui Xin, Yaqi Wang, Xiaotong Jiang, Simin Li, Fengyu Li, Meng Zhou, Christine M. Aikens, Nanfeng Zheng, Hui Shen Engineering molecular rotor-stator ligand architectures on copper nanoclusters for efficient photothermal conversion In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Engineering molecular rotor-stator ligand architectures on copper nanoclusters for efficient photothermal conversion},
author = {Bingzheng Yan and D. Sulalith N. D. Samarasinghe and Jing Sun and Hongwen Deng and Lei Li and Ming-Qiang Qi and Fangming Zhao and Qinghua Xu and Huifang Guo and Xueli Sun and Xuekun Gong and Rong Huo and Mengsi Zhu and Qingyuan Wu and Zhenlang Xie and Chengrui Xin and Yaqi Wang and Xiaotong Jiang and Simin Li and Fengyu Li and Meng Zhou and Christine M. Aikens and Nanfeng Zheng and Hui Shen },
url = {https://www.nature.com/articles/s41467-026-70141-8_reference.pdf},
doi = {10.1038/s41467-026-70141-8},
year = {2026},
date = {2026-03-03},
journal = {Nature Communications},
volume = {17},
abstract = {Copper nanoclusters represent a promising yet underdeveloped frontier in materials science. Here, we propose a general and efficient strategy for enhancing photothermal conversion efficiency through the incorporation of rotor-stator ligand architectures onto copper nanocluster surfaces. As a representative example, we design carboxylate ligands functionalized with adamantane groups to stabilize a [Cu36(4-F-PhS)24(AdmCOO)6(PPh3)4H8]2- nanocluster. In this architecture, the adamantane unit functions as a molecular rotor, while the carboxylate group serves as a molecular stator. The engineered nanocluster achieves a photothermal conversion efficiency of 75%. The adamantane rotors exhibit a lowered rotational energy barrier within the cluster framework, enabling stable and rapid molecular rotation that effectively promotes non-radiative transitions. This mechanism optimizes the conversion of light into thermal energy, enabling the nanocluster to rapidly heat up to 200 °C under 445 nm laser irradiation at a power density of 1.0 W cm-2. The proposed strategy could be applicable to other rotor types, yielding a broad family of copper nanoclusters with enhanced photothermal conversion capabilities and multifunctional potential.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Copper nanoclusters represent a promising yet underdeveloped frontier in materials science. Here, we propose a general and efficient strategy for enhancing photothermal conversion efficiency through the incorporation of rotor-stator ligand architectures onto copper nanocluster surfaces. As a representative example, we design carboxylate ligands functionalized with adamantane groups to stabilize a [Cu36(4-F-PhS)24(AdmCOO)6(PPh3)4H8]2- nanocluster. In this architecture, the adamantane unit functions as a molecular rotor, while the carboxylate group serves as a molecular stator. The engineered nanocluster achieves a photothermal conversion efficiency of 75%. The adamantane rotors exhibit a lowered rotational energy barrier within the cluster framework, enabling stable and rapid molecular rotation that effectively promotes non-radiative transitions. This mechanism optimizes the conversion of light into thermal energy, enabling the nanocluster to rapidly heat up to 200 °C under 445 nm laser irradiation at a power density of 1.0 W cm-2. The proposed strategy could be applicable to other rotor types, yielding a broad family of copper nanoclusters with enhanced photothermal conversion capabilities and multifunctional potential. |
| 288. | | Ryo Ishikawa, Rikuto Kubota, Kazuaki Kawahara, Toshihiro Futazuka, Yuichi Ikuhara, Naoya Shibata 3D dynamic structure of a Pt nanoparticle on SrTiO3 (001) during in-situ heating atomic-resolution ADF STEM imaging In: Nature Communications, vol. 17, no. 1860, 2026, (Active sites in nanocatalysts undergo dynamic structural changes under reaction conditions. Here, the authors use quantitative scanning TEM to reconstruct the 3D dynamic structure of a Pt nanoparticle, identifying excess negative charges accumulated at lower-coordination Pt atomic sites.). @article{nokey,
title = {3D dynamic structure of a Pt nanoparticle on SrTiO3 (001) during in-situ heating atomic-resolution ADF STEM imaging},
author = {Ryo Ishikawa and Rikuto Kubota and Kazuaki Kawahara and Toshihiro Futazuka and Yuichi Ikuhara and Naoya Shibata },
url = {https://www.nature.com/articles/s41467-026-69767-5.pdf},
doi = {10.1038/s41467-026-69767-5},
year = {2026},
date = {2026-02-27},
urldate = {2026-02-27},
journal = {Nature Communications},
volume = {17},
number = {1860},
abstract = {Noble metal nanoparticles supported on semiconducting substrates have long been used in heterogeneous catalysis. A central objective in this field is to identify the active sites for catalysis, which requires precise determination of the 3D atomic structure of nanoparticles on substrates. Furthermore, it has widely accepted that catalytic active sites undergo dynamic structural changes under reaction conditions. However, direct observation of such 3D dynamic structures at the atomic scale, particularly under elevated temperatures and reducing environments, remains a challenge. Here, we present the 3D atomic structure of an epitaxially grown platinum nanoparticle, consisting of 263 Pt atomic sites, supported on an atomically flat SrTiO3 (001) surface, using single-projection atomic-resolution scanning transmission electron microscopy (STEM) combined with quantitative statistical analysis. In-situ heating experiments under reduced atmosphere reveal that the Pt atoms with lower coordination numbers are mobile within the second-nearest-neighbor atomic sites, and we describe the 3D dynamic atomic structure of the Pt nanoparticle by the distribution of partial occupation of Pt atoms. Furthermore, we perform density functional theory calculations based on the experimentally reconstructed 3D atomic structure and find that excess negative charges accumulate at the lower CN atomic sites, which likely serve as catalytically active sites.},
note = {Active sites in nanocatalysts undergo dynamic structural changes under reaction conditions. Here, the authors use quantitative scanning TEM to reconstruct the 3D dynamic structure of a Pt nanoparticle, identifying excess negative charges accumulated at lower-coordination Pt atomic sites.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Noble metal nanoparticles supported on semiconducting substrates have long been used in heterogeneous catalysis. A central objective in this field is to identify the active sites for catalysis, which requires precise determination of the 3D atomic structure of nanoparticles on substrates. Furthermore, it has widely accepted that catalytic active sites undergo dynamic structural changes under reaction conditions. However, direct observation of such 3D dynamic structures at the atomic scale, particularly under elevated temperatures and reducing environments, remains a challenge. Here, we present the 3D atomic structure of an epitaxially grown platinum nanoparticle, consisting of 263 Pt atomic sites, supported on an atomically flat SrTiO3 (001) surface, using single-projection atomic-resolution scanning transmission electron microscopy (STEM) combined with quantitative statistical analysis. In-situ heating experiments under reduced atmosphere reveal that the Pt atoms with lower coordination numbers are mobile within the second-nearest-neighbor atomic sites, and we describe the 3D dynamic atomic structure of the Pt nanoparticle by the distribution of partial occupation of Pt atoms. Furthermore, we perform density functional theory calculations based on the experimentally reconstructed 3D atomic structure and find that excess negative charges accumulate at the lower CN atomic sites, which likely serve as catalytically active sites. |
| 287. | | Yinhan Xu, Meirong Tian, Mengmeng Zhang, Dengwen Hu, Guoqiang Han, Jintao Zhu, Jiangping Xu, Shin-Hyun Kim Dual-responsive coloration of Janus droplets via total internal reflection and interference applied as single-use freezing indicators In: Nature Communications, vol. 17, 2026, (Article in Press). @article{nokey,
title = {Dual-responsive coloration of Janus droplets via total internal reflection and interference applied as single-use freezing indicators},
author = {Yinhan Xu and Meirong Tian and Mengmeng Zhang and Dengwen Hu and Guoqiang Han and Jintao Zhu and Jiangping Xu and Shin-Hyun Kim },
url = {https://www.nature.com/articles/s41467-026-70055-5_reference.pdf},
doi = {10.1038/s41467-026-70055-5},
year = {2026},
date = {2026-02-27},
journal = {Nature Communications},
volume = {17},
abstract = {Responsive optical materials offer real-time visual feedback to external stimuli by converting them into visible light signals, holding great promise in display, sensing, and information encryption. Typically, heterogeneous Janus droplets can exhibit vivid coloration induced by total internal reflection and interference (TIRI) at concave liquid–liquid interfaces. Yet, achieving stimuli-responsive TIRI-based structural colors remains a challenge due to the difficulty in controlling interface curvature and reflective behavior under external stimuli. Here, we report the fabrication of Janus droplets with tunable TIRI structural colors that respond to both surfactant composition and temperature. By tailoring surfactant formulation, the droplet morphology and interface curvature can be adjusted, enabling reversible modulation of structural color appearance, including both iridescent and non-iridescent modes depending on incident light conditions. Furthermore, the structural color can be reversibly or irreversibly switched on/off in response to temperature changes, due to phase transitions that alter light reflection at the concave interface. Owing to their temperature-dependent optical response and real-time visual signaling, these droplets can serve as single-use security labels for monitoring freeze history of freeze-sensitive pharmaceuticals, providing reliable indication of product integrity.},
note = {Article in Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Responsive optical materials offer real-time visual feedback to external stimuli by converting them into visible light signals, holding great promise in display, sensing, and information encryption. Typically, heterogeneous Janus droplets can exhibit vivid coloration induced by total internal reflection and interference (TIRI) at concave liquid–liquid interfaces. Yet, achieving stimuli-responsive TIRI-based structural colors remains a challenge due to the difficulty in controlling interface curvature and reflective behavior under external stimuli. Here, we report the fabrication of Janus droplets with tunable TIRI structural colors that respond to both surfactant composition and temperature. By tailoring surfactant formulation, the droplet morphology and interface curvature can be adjusted, enabling reversible modulation of structural color appearance, including both iridescent and non-iridescent modes depending on incident light conditions. Furthermore, the structural color can be reversibly or irreversibly switched on/off in response to temperature changes, due to phase transitions that alter light reflection at the concave interface. Owing to their temperature-dependent optical response and real-time visual signaling, these droplets can serve as single-use security labels for monitoring freeze history of freeze-sensitive pharmaceuticals, providing reliable indication of product integrity. |
| 286. | | Deping Huang, Zhancheng Li, Yinwu Duan, Xin li, Yongna Zhang, Jiaxing Dong, Guilin Wu, Xiaoxu Huang, Leining Zhang, Feng Ding, Haofei Shi High-throughput chiral copper foils by curved-surface confinement recrystallization In: Nature Communications, vol. 17, no. 2796, 2026, (Preparing chiral metal surfaces on a large scale with high throughput is challenging due to the dependence on chiral templates or molecular precursors. Here, the authors use recrystallization by confinement on curved surfaces to produce chiral copper foils.). @article{nokey,
title = {High-throughput chiral copper foils by curved-surface confinement recrystallization},
author = {Deping Huang and Zhancheng Li and Yinwu Duan and Xin li and Yongna Zhang and Jiaxing Dong and Guilin Wu and Xiaoxu Huang and Leining Zhang and Feng Ding and Haofei Shi },
url = {https://www.nature.com/articles/s41467-026-69862-7.pdf},
doi = {10.1038/s41467-026-69862-7},
year = {2026},
date = {2026-02-20},
urldate = {2026-02-20},
journal = {Nature Communications},
volume = {17},
number = {2796},
abstract = {Chiral metal surfaces play a pivotal role in enantioselective catalysis, sensing, and spintronics, yet their scalable fabrication remains challenging due to a reliance on chiral templates or molecular precursors, which limits both throughput and precise control of crystallographic orientation. Here, we report a high-throughput method for fabricating chiral copper surfaces via curved-surface confinement recrystallization. This approach exploits curvature-driven abnormal grain growth to transform polycrystalline foils into large-area crystals with continuously graded high-index surfaces. Systematic control of the curvature during annealing enabled the creation of a library of chiral copper surfaces, providing high-throughput and surface templates with defined chirality. Through manipulation of the initial crystal orientation and curvature, single crystals with tailored surface orientations can be reached. The intrinsic chirality of these surfaces is confirmed by circular dichroism spectroscopy and model asymmetric reactions. Furthermore, we demonstrate the transfer of chirality to epitaxial two-dimensional materials, exemplified by the growth of chiral graphene. This work provides a scalable platform for producing designer chiral surfaces, enabling future advances in asymmetric catalysis and chiral device engineering.},
note = {Preparing chiral metal surfaces on a large scale with high throughput is challenging due to the dependence on chiral templates or molecular precursors. Here, the authors use recrystallization by confinement on curved surfaces to produce chiral copper foils.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chiral metal surfaces play a pivotal role in enantioselective catalysis, sensing, and spintronics, yet their scalable fabrication remains challenging due to a reliance on chiral templates or molecular precursors, which limits both throughput and precise control of crystallographic orientation. Here, we report a high-throughput method for fabricating chiral copper surfaces via curved-surface confinement recrystallization. This approach exploits curvature-driven abnormal grain growth to transform polycrystalline foils into large-area crystals with continuously graded high-index surfaces. Systematic control of the curvature during annealing enabled the creation of a library of chiral copper surfaces, providing high-throughput and surface templates with defined chirality. Through manipulation of the initial crystal orientation and curvature, single crystals with tailored surface orientations can be reached. The intrinsic chirality of these surfaces is confirmed by circular dichroism spectroscopy and model asymmetric reactions. Furthermore, we demonstrate the transfer of chirality to epitaxial two-dimensional materials, exemplified by the growth of chiral graphene. This work provides a scalable platform for producing designer chiral surfaces, enabling future advances in asymmetric catalysis and chiral device engineering. |
| 285. | | Hairui Lei, Haiyan Qin, Haixin Lei, Yuqing Wang, Jiongzhao Li, Xiaogang Peng Quantum coupling in colloidal homodimers of epitaxially attached CdSe@CdS dot@platelets probed on single-particle level In: Nature Communications, vol. 17, no. 2900, 2026, (Coupled homodimers of quantum dots can provide a platform for investigating quantum phenomena. Here, the authors synthesize homodimers in solution exhibiting two orthogonally polarized photoluminescence modes with two distinct types of biexcitons.). @article{nokey,
title = {Quantum coupling in colloidal homodimers of epitaxially attached CdSe@CdS dot@platelets probed on single-particle level},
author = {Hairui Lei and Haiyan Qin and Haixin Lei and Yuqing Wang and Jiongzhao Li and Xiaogang Peng },
url = {https://www.nature.com/articles/s41467-026-69417-w.pdf},
doi = {10.1038/s41467-026-69417-w},
year = {2026},
date = {2026-02-18},
urldate = {2026-02-18},
journal = {Nature Communications},
volume = {17},
number = {2900},
abstract = {Coupled quantum-dot (QD) dimers can be viewed as diatomic artificial molecules, thus offering a unique platform to study molecular physics and quantum phenomena. However, controlled synthesis of QD dimers has remained an outstanding experimental challenge, given necessary construction of hundreds of lattice bonds at the neck between two QDs with a unique attachment axis. Here, QD homodimers are synthesized in solution with a high yield by the epitaxial attachment of two monodisperse CdSe@CdS dot@platelet QDs along their common <110> axis. The exact construction of hundreds of lattice bonds in the epitaxial neck are enabled by the large (110) side facets with ideal lattice match between two epitaxial QDs and limited ligands passivation, resulting in controlled spacing between two core QDs (core-spacing). Single-particle spectroscopy reveals that the photoluminescence (PL) of single homodimer with a small core-spacing (7.8 nm) is split into two room-temperature resolvable peaks, consistent with strongly-split bonding and antibonding electron orbitals of “artificial diatomic molecules”. The two peaks of the split PL are orthogonally-polarized, and two types of molecular bi-excitons are observed for the homodimer. These advancements lay a viable foundation for controllable construction of “artificial molecules” in solution.},
note = {Coupled homodimers of quantum dots can provide a platform for investigating quantum phenomena. Here, the authors synthesize homodimers in solution exhibiting two orthogonally polarized photoluminescence modes with two distinct types of biexcitons.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coupled quantum-dot (QD) dimers can be viewed as diatomic artificial molecules, thus offering a unique platform to study molecular physics and quantum phenomena. However, controlled synthesis of QD dimers has remained an outstanding experimental challenge, given necessary construction of hundreds of lattice bonds at the neck between two QDs with a unique attachment axis. Here, QD homodimers are synthesized in solution with a high yield by the epitaxial attachment of two monodisperse CdSe@CdS dot@platelet QDs along their common <110> axis. The exact construction of hundreds of lattice bonds in the epitaxial neck are enabled by the large (110) side facets with ideal lattice match between two epitaxial QDs and limited ligands passivation, resulting in controlled spacing between two core QDs (core-spacing). Single-particle spectroscopy reveals that the photoluminescence (PL) of single homodimer with a small core-spacing (7.8 nm) is split into two room-temperature resolvable peaks, consistent with strongly-split bonding and antibonding electron orbitals of “artificial diatomic molecules”. The two peaks of the split PL are orthogonally-polarized, and two types of molecular bi-excitons are observed for the homodimer. These advancements lay a viable foundation for controllable construction of “artificial molecules” in solution. |
| 284. | | Hao-Jun Qin, Rui-Jing Sun, Jia-Jun Liu, Wen-Ao Liao, Dao-Bo Wang, Jiang Yu, Tian-Hao Leng, Chao-Fei Liu, Wen-hao Zhang, Ying-Shuang Fu Molecular electronic chirality in copper phthalocyanine induced via twisted π-π stacking on bilayer graphene In: Nature Communications, vol. 17, no. 3130, 2026, (Electronic chirality is a fascinating but still poorly understood phenomenon. Here, the authors show that CuPc molecules on bilayer graphene exhibit electronic chirality driven by twisted π–π interactions, which can be reversibly switched using an STM tip.). @article{nokey,
title = {Molecular electronic chirality in copper phthalocyanine induced via twisted π-π stacking on bilayer graphene},
author = {Hao-Jun Qin and Rui-Jing Sun and Jia-Jun Liu and Wen-Ao Liao and Dao-Bo Wang and Jiang Yu and Tian-Hao Leng and Chao-Fei Liu and Wen-hao Zhang and Ying-Shuang Fu },
url = {https://www.nature.com/articles/s41467-026-69713-5.pdf},
doi = {10.1038/s41467-026-69713-5},
year = {2026},
date = {2026-02-16},
urldate = {2026-02-16},
journal = {Nature Communications},
volume = {17},
number = {3130},
abstract = {Electronic chirality within single molecules constitutes an intriguing phenomenon in quantum chemistry, whose inducing mechanism however remains underexplored. Here, we report a distinct formation mechanism for electronic chirality in CuPc molecules adsorbed on bilayer graphene on highly oriented pyrolytic graphite, as incurred via twisted π-π stacking. Scanning tunneling microscopy measurements unveil that CuPc molecules exhibit prominent chirality in morphology at low biases, but restore their D4h symmetry at large biases, demonstrating the chirality is electronic origin. With tip manipulations, the two enantiomers of CuPc can be reversibly switched. Density functional theory calculations reveal that the electronic chirality arises from π-π hybridization between CuPc and graphene, leading to asymmetric charge distribution. The chiral configuration is determined by adsorption sites and rotation angles relative to graphene, in agreement with experimental observations. This work uncovers a π-π hybridization mechanism for driving electronic chirality, providing a platform for designing chiral molecular electronic devices.},
note = {Electronic chirality is a fascinating but still poorly understood phenomenon. Here, the authors show that CuPc molecules on bilayer graphene exhibit electronic chirality driven by twisted π–π interactions, which can be reversibly switched using an STM tip.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Electronic chirality within single molecules constitutes an intriguing phenomenon in quantum chemistry, whose inducing mechanism however remains underexplored. Here, we report a distinct formation mechanism for electronic chirality in CuPc molecules adsorbed on bilayer graphene on highly oriented pyrolytic graphite, as incurred via twisted π-π stacking. Scanning tunneling microscopy measurements unveil that CuPc molecules exhibit prominent chirality in morphology at low biases, but restore their D4h symmetry at large biases, demonstrating the chirality is electronic origin. With tip manipulations, the two enantiomers of CuPc can be reversibly switched. Density functional theory calculations reveal that the electronic chirality arises from π-π hybridization between CuPc and graphene, leading to asymmetric charge distribution. The chiral configuration is determined by adsorption sites and rotation angles relative to graphene, in agreement with experimental observations. This work uncovers a π-π hybridization mechanism for driving electronic chirality, providing a platform for designing chiral molecular electronic devices. |
| 283. | | Ekaterina Salikhova, Alf Mews, Hendrik Schlicke, Jan Steffen Niehaus Colloidal synthesis of large near-bulk InAs quantum dots through seeded and seedless growth using cluster precursors In: Nature Communications, vol. 17, no. 1700, 2026, (Colloidal quantum dots are promising, low-cost materials for infrared technologies, but rarely comply with the EU directive on the restriction of the use of certain hazardous substances (RoHS). Here, the authors present the synthesis of large, near-bulk RoHS-compliant quantum dots, operating deep in the infrared.). @article{nokey,
title = {Colloidal synthesis of large near-bulk InAs quantum dots through seeded and seedless growth using cluster precursors},
author = {Ekaterina Salikhova and Alf Mews and Hendrik Schlicke and Jan Steffen Niehaus },
url = {https://www.nature.com/articles/s41467-026-69409-w.pdf},
doi = {10.1038/s41467-026-69409-w},
year = {2026},
date = {2026-02-12},
journal = {Nature Communications},
volume = {17},
number = {1700},
abstract = {Quantum dots are high-potential materials for a wide range of applications, particularly in the infrared spectral range, including telecommunications, security, sensing, photovoltaics, and bioimaging. The size and chemical composition of semiconductor quantum dots determine their optoelectronic properties. While II-VI and IV-VI quantum dots are well-studied, the colloidal synthesis of infrared-active III-V quantum dots such as InAs, which are compliant with the European Union directive for restriction of hazardous substances, remains underdeveloped. In particular, the synthesis of larger InAs quantum dots is challenging due to the strong covalent character of In–As bonding, limited control over precursor reactivity, and complex growth pathways. Here we show the synthesis of large, near-bulk InAs quantum dots using atomic clusters as precursors. All nanostructures are synthesized from ‘green’ commercially available precursors. These results extend the accessible size regime of colloidal InAs nanoparticles to diameters approaching 40 nm and establish a platform for their use in infrared technologies.},
note = {Colloidal quantum dots are promising, low-cost materials for infrared technologies, but rarely comply with the EU directive on the restriction of the use of certain hazardous substances (RoHS). Here, the authors present the synthesis of large, near-bulk RoHS-compliant quantum dots, operating deep in the infrared.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Quantum dots are high-potential materials for a wide range of applications, particularly in the infrared spectral range, including telecommunications, security, sensing, photovoltaics, and bioimaging. The size and chemical composition of semiconductor quantum dots determine their optoelectronic properties. While II-VI and IV-VI quantum dots are well-studied, the colloidal synthesis of infrared-active III-V quantum dots such as InAs, which are compliant with the European Union directive for restriction of hazardous substances, remains underdeveloped. In particular, the synthesis of larger InAs quantum dots is challenging due to the strong covalent character of In–As bonding, limited control over precursor reactivity, and complex growth pathways. Here we show the synthesis of large, near-bulk InAs quantum dots using atomic clusters as precursors. All nanostructures are synthesized from ‘green’ commercially available precursors. These results extend the accessible size regime of colloidal InAs nanoparticles to diameters approaching 40 nm and establish a platform for their use in infrared technologies. |
| 282. | | Xuelei Pan, Davide Bernardo Preso, Qian Liu, Lucia Mullings, Mohamad Salimi, Yi Qin, Pankaj S. Sinhmar, James Kwan Mechanistic insights into the non-equilibrium thermodynamics of nitrogen fixation via acoustic cavitation In: Nature Communications, vol. 17, no. 2682, 2026, (Industrial nitrogen fixation relies on energy-intensive processes. Here, the authors provide mechanistic insights on the transient conditions in acoustic cavitation that activate nitrogen and form nitrogen products in the absence of catalysts.). @article{nokey,
title = {Mechanistic insights into the non-equilibrium thermodynamics of nitrogen fixation via acoustic cavitation},
author = {Xuelei Pan and Davide Bernardo Preso and Qian Liu and Lucia Mullings and Mohamad Salimi and Yi Qin and Pankaj S. Sinhmar and James Kwan },
url = {https://www.nature.com/articles/s41467-026-69466-1.pdf},
doi = {10.1038/s41467-026-69466-1},
year = {2026},
date = {2026-02-12},
urldate = {2026-02-12},
journal = {Nature Communications},
volume = {17},
number = {2682},
abstract = {Non-equilibrium reaction environments offer a route to bypass the thermodynamic constraints that limit conventional nitrogen fixation, yet such conditions remain inaccessible in traditional thermal systems. Here, we show that rapid activation-quenching chemistry inside cavitation bubbles provides a viable non‑equilibrium pathway for nitrogen fixation. The violent collapse of ultrasound-driven bubbles generates an intense temperature pulse that enables direct nitrogen activation and subsequent redox chemistry within a transient gas phase microreactor. Nitrogen-containing products are produced with tuneable rates and selectivity controlled by feed gas composition, cavitation dynamics, and solution properties. Introduced cavitation nuclei lower the cavitation threshold and improve collapse reproducibility, while noble‑gas doping modulates collapse temperatures and shifts nitrate-nitrite distributions through enhancing the involvement of water‑derived species. Isotopic labelling and single‑bubble modelling indicate that nitrogen reaction proceeds predominantly through gas‑phase pathways during collapse, which can be described by a dynamic thermodynamic model within a temperature pulse. These findings establish cavitation‑driven non-equilibrium thermal cycling as a distinct mechanism for nitrogen fixation and underscore the broader potential of transient thermal microenvironments for chemical synthesis.},
note = {Industrial nitrogen fixation relies on energy-intensive processes. Here, the authors provide mechanistic insights on the transient conditions in acoustic cavitation that activate nitrogen and form nitrogen products in the absence of catalysts.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Non-equilibrium reaction environments offer a route to bypass the thermodynamic constraints that limit conventional nitrogen fixation, yet such conditions remain inaccessible in traditional thermal systems. Here, we show that rapid activation-quenching chemistry inside cavitation bubbles provides a viable non‑equilibrium pathway for nitrogen fixation. The violent collapse of ultrasound-driven bubbles generates an intense temperature pulse that enables direct nitrogen activation and subsequent redox chemistry within a transient gas phase microreactor. Nitrogen-containing products are produced with tuneable rates and selectivity controlled by feed gas composition, cavitation dynamics, and solution properties. Introduced cavitation nuclei lower the cavitation threshold and improve collapse reproducibility, while noble‑gas doping modulates collapse temperatures and shifts nitrate-nitrite distributions through enhancing the involvement of water‑derived species. Isotopic labelling and single‑bubble modelling indicate that nitrogen reaction proceeds predominantly through gas‑phase pathways during collapse, which can be described by a dynamic thermodynamic model within a temperature pulse. These findings establish cavitation‑driven non-equilibrium thermal cycling as a distinct mechanism for nitrogen fixation and underscore the broader potential of transient thermal microenvironments for chemical synthesis. |
| 281. | | Junhong Wu, Hao Li, Jialu Wang, Xiaorui Yang, Tianyong Zhang, Bin Li, Shuang Jiang Mechanochemical engineering of chiroptical properties in indium-based chiral metal halides by grinding In: Nature Communications, vol. 17, no. 2619, 2026, (Nanomaterials with chiral photoluminescence are promising candidates for advanced optical technologies. Here, the authors use mechsanochemical processing to tune the emission of circularly polarized light and enhance the nonlinear optical response in indium-based halides.). @article{nokey,
title = {Mechanochemical engineering of chiroptical properties in indium-based chiral metal halides by grinding},
author = {Junhong Wu and Hao Li and Jialu Wang and Xiaorui Yang and Tianyong Zhang and Bin Li and Shuang Jiang },
url = {https://www.nature.com/articles/s41467-026-69353-9.pdf},
doi = {10.1038/s41467-026-69353-9},
year = {2026},
date = {2026-02-11},
urldate = {2026-02-11},
journal = {Nature Communications},
volume = {17},
number = {2619},
abstract = {Circularly polarized luminescence is crucial for optoelectronics, bioimaging, and three-dimensional display, yet most of current materials suffer from complex synthesis and limited tunability. Herein, we show the regulation of chiroptical properties in metal halides through mechanochemical engineering. Specifically, phosphorescent indium-based chiral metal halides exhibit blue circularly polarized luminescence, along with antimony-doped indium-based metal halides emit orange circularly polarized luminescence. A grinding strategy using bromide salts like potassium bromide induces bright yellow fluorescence and enables versatile circularly polarized luminescence modulation. When antimony-doped indium-based metal halides are ground with five different bromide salts, it exhibits intriguing and tunable properties: (i) enhanced circularly polarized luminescence, with a luminescence dissymmetry factor value up to 10⁻²; (ii) inversion of the circularly polarized luminescence signal; (iii) generation of near-infrared circularly polarized luminescence with a substantial Stokes shift of 370 nm; and most notably, (iv) a 29.71-fold improvement in second-harmonic generation efficiency. This approach also realizes applications in circularly polarized light-emitting diodes.},
note = {Nanomaterials with chiral photoluminescence are promising candidates for advanced optical technologies. Here, the authors use mechsanochemical processing to tune the emission of circularly polarized light and enhance the nonlinear optical response in indium-based halides.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Circularly polarized luminescence is crucial for optoelectronics, bioimaging, and three-dimensional display, yet most of current materials suffer from complex synthesis and limited tunability. Herein, we show the regulation of chiroptical properties in metal halides through mechanochemical engineering. Specifically, phosphorescent indium-based chiral metal halides exhibit blue circularly polarized luminescence, along with antimony-doped indium-based metal halides emit orange circularly polarized luminescence. A grinding strategy using bromide salts like potassium bromide induces bright yellow fluorescence and enables versatile circularly polarized luminescence modulation. When antimony-doped indium-based metal halides are ground with five different bromide salts, it exhibits intriguing and tunable properties: (i) enhanced circularly polarized luminescence, with a luminescence dissymmetry factor value up to 10⁻²; (ii) inversion of the circularly polarized luminescence signal; (iii) generation of near-infrared circularly polarized luminescence with a substantial Stokes shift of 370 nm; and most notably, (iv) a 29.71-fold improvement in second-harmonic generation efficiency. This approach also realizes applications in circularly polarized light-emitting diodes. |
| 280. | | Liu-Liu Shen, Gui-Rong Zhang, Weiwei Zhang, Wen-Tao Zheng, Mingjian Wu, Erdmann Spiecker, Donghai Mei, Bastian J. M. Etzold Synthesis of 2D amorphous carbons via energy-autonomous carbonization of polyaniline upon decomposition of HClO₄ In: Nature Communications, vol. 17, no. 2485, 2026, (Conventional carbon synthesis is energy intensive. Here, the authors introduce an energy-autonomous pathway using polyaniline-HClO4 composites. This rapid reaction yields carbon nanosheets with tunable active sites for electrocatalysis.). @article{nokey,
title = {Synthesis of 2D amorphous carbons via energy-autonomous carbonization of polyaniline upon decomposition of HClO₄},
author = {Liu-Liu Shen and Gui-Rong Zhang and Weiwei Zhang and Wen-Tao Zheng and Mingjian Wu and Erdmann Spiecker and Donghai Mei and Bastian J. M. Etzold },
url = {https://www.nature.com/articles/s41467-026-69314-2.pdf},
doi = {10.1038/s41467-026-69314-2},
year = {2026},
date = {2026-02-07},
urldate = {2026-02-07},
journal = {Nature Communications},
volume = {17},
number = {2485},
abstract = {Despite centuries of advancement, the synthesis of carbon materials remains heavily reliant on energy-intensive thermal processes. Conventional methods require external heating for prolonged periods to overcome high energy barriers, posing challenges for sustainable large-scale production. Here we show an energy-autonomous synthesis pathway that utilizes the intrinsic chemical energy stored within a polyaniline-HClO4 composite. Triggered by mild thermal, microwave, or mechanical stimulation, the precursor undergoes a rapid exothermic self-propagation driven by the explosive decomposition of perchlorate species. This single-step process, completed in ≈0.4 s, simultaneously generates intense localized heat and a massive volume of gas, which forcibly exfoliates and carbonizes the polymer into interconnected 2D amorphous carbon nanosheets. We demonstrate that this energy-efficient method achieves carbon conversion efficiencies comparable to traditional pyrolysis. Furthermore, the reaction intensity is precisely tunable via the precursor water content, ensuring potential for safe industrial scale-up. This approach also enables the atomic-level incorporation of transition metals, creating a versatile platform for the design of catalysts for oxygen and carbon dioxide reduction reactions. This work provides a scalable, energy-autonomous pathway for carbon synthesis and offers a platform for the precise construction of catalytic architectures.},
note = {Conventional carbon synthesis is energy intensive. Here, the authors introduce an energy-autonomous pathway using polyaniline-HClO4 composites. This rapid reaction yields carbon nanosheets with tunable active sites for electrocatalysis.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Despite centuries of advancement, the synthesis of carbon materials remains heavily reliant on energy-intensive thermal processes. Conventional methods require external heating for prolonged periods to overcome high energy barriers, posing challenges for sustainable large-scale production. Here we show an energy-autonomous synthesis pathway that utilizes the intrinsic chemical energy stored within a polyaniline-HClO4 composite. Triggered by mild thermal, microwave, or mechanical stimulation, the precursor undergoes a rapid exothermic self-propagation driven by the explosive decomposition of perchlorate species. This single-step process, completed in ≈0.4 s, simultaneously generates intense localized heat and a massive volume of gas, which forcibly exfoliates and carbonizes the polymer into interconnected 2D amorphous carbon nanosheets. We demonstrate that this energy-efficient method achieves carbon conversion efficiencies comparable to traditional pyrolysis. Furthermore, the reaction intensity is precisely tunable via the precursor water content, ensuring potential for safe industrial scale-up. This approach also enables the atomic-level incorporation of transition metals, creating a versatile platform for the design of catalysts for oxygen and carbon dioxide reduction reactions. This work provides a scalable, energy-autonomous pathway for carbon synthesis and offers a platform for the precise construction of catalytic architectures. |
| 279. | | Zhiwen Zhu, Qi Huang, Tairan Yang, Hao Jiang, Shaoxuan Yuan, Juan Xiang, Liangliang Cai, Qiang Sun Deep learning drives autonomous molecular reactions with single-bond selectivity in tetra-brominated porphyrins on Au(111) In: Nature Communications, vol. 17, no. 2348, 2026, (Controlling chemical reactions one bond at a time is a challenge in nanoscience. Here, the authors present an AI-controlled STM system that autonomously performs multistep, bond-selective reactions on individual molecules with high precision and reproducibility.). @article{nokey,
title = {Deep learning drives autonomous molecular reactions with single-bond selectivity in tetra-brominated porphyrins on Au(111)},
author = {Zhiwen Zhu and Qi Huang and Tairan Yang and Hao Jiang and Shaoxuan Yuan and Juan Xiang and Liangliang Cai and Qiang Sun },
url = {https://www.nature.com/articles/s41467-026-69080-1.pdf},
doi = {10.1038/s41467-026-69080-1},
year = {2026},
date = {2026-02-04},
urldate = {2026-02-04},
journal = {Nature Communications},
volume = {17},
number = {2348},
abstract = {The pursuit of autonomous chemical transformations with single-bond precision represents a central challenge in molecular nanoscience. While scanning tunneling microscopy (STM) enables site-specific reactions by directly engaging individual atoms and bonds, conventional approaches rely on expert intervention and lack reproducibility and scalability. Here we introduce a deep learning-based strategy that autonomously executes multi-step, bond-selective transformations. Our system integrates computer vision for molecular recognition, neural networks for bond-state classification, and deep reinforcement learning for closed-loop optimization of activation parameters. As a proof of concept, we demonstrate the selective dissociation of C-Br bonds in a tetra-brominated porphyrin on Au(111). Importantly, the approach extends beyond single-bond events, enabling programmed multi-step sequences including four distinct pathways with high fidelity. By advancing from isolated, human-directed manipulations to fully autonomous, data-driven reaction control, this platform establishes a paradigm for intelligent single-molecule chemistry. It provides a generalizable framework for on-surface synthesis, where adaptive agents orchestrate molecular transformations with a level of precision and scalability unattainable by manual approaches.},
note = {Controlling chemical reactions one bond at a time is a challenge in nanoscience. Here, the authors present an AI-controlled STM system that autonomously performs multistep, bond-selective reactions on individual molecules with high precision and reproducibility.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The pursuit of autonomous chemical transformations with single-bond precision represents a central challenge in molecular nanoscience. While scanning tunneling microscopy (STM) enables site-specific reactions by directly engaging individual atoms and bonds, conventional approaches rely on expert intervention and lack reproducibility and scalability. Here we introduce a deep learning-based strategy that autonomously executes multi-step, bond-selective transformations. Our system integrates computer vision for molecular recognition, neural networks for bond-state classification, and deep reinforcement learning for closed-loop optimization of activation parameters. As a proof of concept, we demonstrate the selective dissociation of C-Br bonds in a tetra-brominated porphyrin on Au(111). Importantly, the approach extends beyond single-bond events, enabling programmed multi-step sequences including four distinct pathways with high fidelity. By advancing from isolated, human-directed manipulations to fully autonomous, data-driven reaction control, this platform establishes a paradigm for intelligent single-molecule chemistry. It provides a generalizable framework for on-surface synthesis, where adaptive agents orchestrate molecular transformations with a level of precision and scalability unattainable by manual approaches. |
| 278. | | Lalith Krishna Samanth Bonagiri, Diana M. Arvelo, Fujia Zhao, Jaehyeon Kim, Qian Ai, Shan Zhou, Kaustubh S. Panse, Ricardo Garcia, Yingjie Zhang Probing the molecular structure at graphite–water interfaces by correlating 3D-AFM and SHINERS In: Nature Communications, vol. 17, no. 2230, 2026, (Interfacial water is of crucial importance in biology and engineering. Here, the authors identify structural transitions of interfacial water at graphite surfaces under varying electric fields and environmental conditions.). @article{nokey,
title = {Probing the molecular structure at graphite–water interfaces by correlating 3D-AFM and SHINERS},
author = {Lalith Krishna Samanth Bonagiri and Diana M. Arvelo and Fujia Zhao and Jaehyeon Kim and Qian Ai and Shan Zhou and Kaustubh S. Panse and Ricardo Garcia and Yingjie Zhang },
url = {https://www.nature.com/articles/s41467-026-68667-y.pdf},
doi = {10.1038/s41467-026-68667-y},
year = {2026},
date = {2026-01-31},
urldate = {2026-01-31},
journal = {Nature Communications},
volume = {17},
number = {2230},
abstract = {Water at solid surfaces is key for many processes ranging from biological signal transduction to membrane separation and renewable energy conversion. However, under realistic conditions, which often include environmental and surface charge variations, the interfacial water structure remains elusive. Here we overcome this limit by combining three-dimensional atomic force microscopy (3D-AFM) and interface-sensitive shell-isolated nanoparticle enhanced Raman spectroscopy (SHINERS) to characterize the graphite–water interfacial structure in situ. Through correlative analysis of the spatial liquid density maps and vibrational peaks within ≈2 nm of the graphite surface, we find the existence of two interfacial configurations at open circuit potential, a transient state where pristine water exhibits strong hydrogen bond (H-bond) breaking effects, and a steady state with hydrocarbons dominating the interface and weak H-bond breaking in the surrounding water. At sufficiently negative potentials, both states transition into a stable structure featuring pristine water with a broader distribution of H-bond configurations. Our three-state model resolves many long-standing controversies on interfacial water structure.},
note = {Interfacial water is of crucial importance in biology and engineering. Here, the authors identify structural transitions of interfacial water at graphite surfaces under varying electric fields and environmental conditions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Water at solid surfaces is key for many processes ranging from biological signal transduction to membrane separation and renewable energy conversion. However, under realistic conditions, which often include environmental and surface charge variations, the interfacial water structure remains elusive. Here we overcome this limit by combining three-dimensional atomic force microscopy (3D-AFM) and interface-sensitive shell-isolated nanoparticle enhanced Raman spectroscopy (SHINERS) to characterize the graphite–water interfacial structure in situ. Through correlative analysis of the spatial liquid density maps and vibrational peaks within ≈2 nm of the graphite surface, we find the existence of two interfacial configurations at open circuit potential, a transient state where pristine water exhibits strong hydrogen bond (H-bond) breaking effects, and a steady state with hydrocarbons dominating the interface and weak H-bond breaking in the surrounding water. At sufficiently negative potentials, both states transition into a stable structure featuring pristine water with a broader distribution of H-bond configurations. Our three-state model resolves many long-standing controversies on interfacial water structure. |
| 277. | | Hai-Zhen Yu, Rui-Lin Han, Dingwei Chu, Yuanzhi Li, Xiao-Ru Dong, Yang Zhang, Li Wang, Yuzhi Song, Chuan-Kui Wang, Zhen Xie, Sai Duan Theoretical prediction for monitoring Jahn-Teller vibrational evolution using real-space tip-enhanced Raman imaging In: Nature Communications, vol. 17, no. 1132, 2026, (Jahn-Teller distortions alter molecular structures, but are usually only probed on the basis of electronic signatures. Here, the authors demonstrate that tip-enhanced Raman imaging in real space can map the associated vibrational changes in a single molecule.). @article{nokey,
title = {Theoretical prediction for monitoring Jahn-Teller vibrational evolution using real-space tip-enhanced Raman imaging},
author = {Hai-Zhen Yu and Rui-Lin Han and Dingwei Chu and Yuanzhi Li and Xiao-Ru Dong and Yang Zhang and Li Wang and Yuzhi Song and Chuan-Kui Wang and Zhen Xie and Sai Duan },
url = {https://www.nature.com/articles/s41467-025-67894-z.pdf},
doi = {10.1038/s41467-025-67894-z},
year = {2026},
date = {2026-01-09},
urldate = {2026-01-09},
journal = {Nature Communications},
volume = {17},
number = {1132},
abstract = {The Jahn-Teller effect (JTE) reduces the geometrical symmetry of a system with degenerate electronic states via vibronic coupling, playing a pivotal role in molecular and condensed systems. However, due to the intrinsic limitations of conventional techniques, only the electronic evolution in JTE can be measured. Herein, we theoretically propose that vibrational resolved tip-enhanced Raman scattering images can visualize the vibrational evolutions in JTE in real space. Taking an experimentally accessible single zinc phthalocyanine (ZnPc) molecule as a proof-of-principle example, not only the degenerate vibrational splitting but also the overlooked vibration mixing caused by the JTE in its anionic form can be characterized by Raman images. Leveraging Raman images, the controllable configuration of JTE distortion with partial isotopic substitution can be further identified. These findings establish a practical protocol to monitor the detailed vibrational evolutions when a single molecule experiences JTE, providing a pathway for visualization of spontaneous symmetry breaking in molecular and solid-state systems.},
note = {Jahn-Teller distortions alter molecular structures, but are usually only probed on the basis of electronic signatures. Here, the authors demonstrate that tip-enhanced Raman imaging in real space can map the associated vibrational changes in a single molecule.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The Jahn-Teller effect (JTE) reduces the geometrical symmetry of a system with degenerate electronic states via vibronic coupling, playing a pivotal role in molecular and condensed systems. However, due to the intrinsic limitations of conventional techniques, only the electronic evolution in JTE can be measured. Herein, we theoretically propose that vibrational resolved tip-enhanced Raman scattering images can visualize the vibrational evolutions in JTE in real space. Taking an experimentally accessible single zinc phthalocyanine (ZnPc) molecule as a proof-of-principle example, not only the degenerate vibrational splitting but also the overlooked vibration mixing caused by the JTE in its anionic form can be characterized by Raman images. Leveraging Raman images, the controllable configuration of JTE distortion with partial isotopic substitution can be further identified. These findings establish a practical protocol to monitor the detailed vibrational evolutions when a single molecule experiences JTE, providing a pathway for visualization of spontaneous symmetry breaking in molecular and solid-state systems. |
| 276. | | Yunluo Wang, Hang Yang, Wenxuan Yao, Jie Chen, Haoming Qin, Yuge Cao, Xuchang He, Jianghua Wu, Hao Gu, Ruifeng Liu, Yan Yang, Zesen Gao, Futing Sun, Tianshuo Zhang, Tianrui Zhou, Yuqiang Fang, Qinhua Wei, Jingshan Hou, Yongzheng Fang, Yihui He, Xiang Li, Yi-Yang Sun, Lianjun Wang, Wan Jiang, Dong Tu, Guogang Li, Fuqiang Huang, Haijie Chen Mechanistic insight into the Young′s modulus threshold for fracto-mechanoluminescence in manganese halides In: Nature Communications, vol. 17, no. 1154, 2026, (Fracto-mechanoluminescence is the emission of light from fracturing solids. Here, the authors show that this emission in Mn halides arises from elastic stiffness, electromechanical coupling, and trap states that activate Mn2+ d−d transitions.). @article{nokey,
title = {Mechanistic insight into the Young′s modulus threshold for fracto-mechanoluminescence in manganese halides},
author = {Yunluo Wang and Hang Yang and Wenxuan Yao and Jie Chen and Haoming Qin and Yuge Cao and Xuchang He and Jianghua Wu and Hao Gu and Ruifeng Liu and Yan Yang and Zesen Gao and Futing Sun and Tianshuo Zhang and Tianrui Zhou and Yuqiang Fang and Qinhua Wei and Jingshan Hou and Yongzheng Fang and Yihui He and Xiang Li and Yi-Yang Sun and Lianjun Wang and Wan Jiang and Dong Tu and Guogang Li and Fuqiang Huang and Haijie Chen},
url = {https://www.nature.com/articles/s41467-025-67914-y.pdf},
doi = {10.1038/s41467-025-67914-y},
year = {2026},
date = {2026-01-09},
urldate = {2026-01-09},
journal = {Nature Communications},
volume = {17},
number = {1154},
abstract = {Fracto-mechanoluminescence (FML), a subtype of mechanoluminescence, is the phenomenon of light emission triggered by the fracturing of solids under mechanical stimuli. Although many materials have been reported with FML, the underlying mechanism remains unclear, leveraging the fundamental prerequisites and design principles to achieve FML remain elusive. In this study, we systematically investigate a series of Mn halides and find that 12 out of 18 compounds exhibit bright and clearly detectable FML. Here we show that the occurrence of FML arises from the synergistic interplay among the crystal′s elastic stiffness, local electromechanical coupling, and trap states, which collectively activate Mn2+ d−d transitions upon fracture. The enhancement of FML intensity is primarily governed by the enlargement of the effective fracture area, whereas the Young′s modulus determines the fracture threshold and the tolerable stress range of crystals. Additionally, as-explored Mn halides exhibit improved X-ray imaging capabilities, which are further integrated into radiation warning and damage detection devices.},
note = {Fracto-mechanoluminescence is the emission of light from fracturing solids. Here, the authors show that this emission in Mn halides arises from elastic stiffness, electromechanical coupling, and trap states that activate Mn2+ d−d transitions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fracto-mechanoluminescence (FML), a subtype of mechanoluminescence, is the phenomenon of light emission triggered by the fracturing of solids under mechanical stimuli. Although many materials have been reported with FML, the underlying mechanism remains unclear, leveraging the fundamental prerequisites and design principles to achieve FML remain elusive. In this study, we systematically investigate a series of Mn halides and find that 12 out of 18 compounds exhibit bright and clearly detectable FML. Here we show that the occurrence of FML arises from the synergistic interplay among the crystal′s elastic stiffness, local electromechanical coupling, and trap states, which collectively activate Mn2+ d−d transitions upon fracture. The enhancement of FML intensity is primarily governed by the enlargement of the effective fracture area, whereas the Young′s modulus determines the fracture threshold and the tolerable stress range of crystals. Additionally, as-explored Mn halides exhibit improved X-ray imaging capabilities, which are further integrated into radiation warning and damage detection devices. |
| 275. | | Maryam Sabooni Asre Hazer, Sami Malola, Hannu Häkkinen Thermal dynamics and coalescence of Au144(SR)60 clusters from a machine-learned potential In: Nature Communications, vol. 17, no. 971, 2025, (Understanding the dynamic behavior of metal clusters at high temperatures is challenging due to a lack of theoretical knowledge. Here, the authors use a machine-learned potential for thiolate-protected gold clusters to reveal atom-scale mechanisms of cluster disordering and cluster-cluster reactions.). @article{nokey,
title = {Thermal dynamics and coalescence of Au144(SR)60 clusters from a machine-learned potential},
author = {Maryam Sabooni Asre Hazer and Sami Malola and Hannu Häkkinen },
url = {https://www.nature.com/articles/s41467-025-67700-w.pdf},
doi = {10.1038/s41467-025-67700-w},
year = {2025},
date = {2025-12-18},
urldate = {2025-12-18},
journal = {Nature Communications},
volume = {17},
number = {971},
abstract = {Ligand-protected metal nanoclusters have gained significant attention due to their diverse applications in catalysis, bioimaging, and nanomedicine. While their ground-state electronic structure and optical properties have been extensively studied via density functional theory (DFT) methods, theoretical insights into their dynamic behavior, particularly for larger clusters, remain scarce due to the prohibitive numerical cost of long-timescale simulations using forces calculated from DFT. Here we investigate, using molecular dynamics (MD) simulations up to 0.12 μs timescale, thermal dynamics of the well-known Au144(SR)60 at 300-550 K using the recently parametrized atomic cluster expansion (ACE) potential, trained from DFT data for thiolate-protected gold clusters. Our findings reveal that thermal effects induce increased mobility in a layer-by-layer fashion, leading to formation of polymeric gold-thiolate units and rings which may fragment from the cluster at high temperatures. The remaining smaller clusters resemble experimentally observed cluster compositions. Close interaction of two Au144(SR)60 clusters leads to coalescence, resulting in a cluster composition and structure of the inner metal core close to ones identified in previous experiments. This work reveals mechanisms for thermal effects in ligand-protected gold clusters and larger nanoparticles that are instrumental for understanding their catalytic activity and inter-particle reactions at the atomistic level.},
note = {Understanding the dynamic behavior of metal clusters at high temperatures is challenging due to a lack of theoretical knowledge. Here, the authors use a machine-learned potential for thiolate-protected gold clusters to reveal atom-scale mechanisms of cluster disordering and cluster-cluster reactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ligand-protected metal nanoclusters have gained significant attention due to their diverse applications in catalysis, bioimaging, and nanomedicine. While their ground-state electronic structure and optical properties have been extensively studied via density functional theory (DFT) methods, theoretical insights into their dynamic behavior, particularly for larger clusters, remain scarce due to the prohibitive numerical cost of long-timescale simulations using forces calculated from DFT. Here we investigate, using molecular dynamics (MD) simulations up to 0.12 μs timescale, thermal dynamics of the well-known Au144(SR)60 at 300-550 K using the recently parametrized atomic cluster expansion (ACE) potential, trained from DFT data for thiolate-protected gold clusters. Our findings reveal that thermal effects induce increased mobility in a layer-by-layer fashion, leading to formation of polymeric gold-thiolate units and rings which may fragment from the cluster at high temperatures. The remaining smaller clusters resemble experimentally observed cluster compositions. Close interaction of two Au144(SR)60 clusters leads to coalescence, resulting in a cluster composition and structure of the inner metal core close to ones identified in previous experiments. This work reveals mechanisms for thermal effects in ligand-protected gold clusters and larger nanoparticles that are instrumental for understanding their catalytic activity and inter-particle reactions at the atomistic level. |
| 274. | | Sebastian T. Mergelsberg, Hongyou Fan, James J. De Yoreo, Carolyn I. Pearce, Kevin M. Rosso, Xin Zhang Mesocrystal growth through oriented sliding and attachment of nanoplates In: Nature Communications, vol. 16, no. 11240, 2025, (Oriented growth is an important pathway for crystal growth. Here, the authors show that gibbsite nanoplates form mesocrystals through directed sliding and staggered stacking, as demonstrated by in situ microscopy and molecular simulations.). @article{nokey,
title = {Mesocrystal growth through oriented sliding and attachment of nanoplates},
author = {Sebastian T. Mergelsberg and Hongyou Fan and James J. De Yoreo and Carolyn I. Pearce and Kevin M. Rosso and Xin Zhang },
url = {https://www.nature.com/articles/s41467-025-64852-7.pdf},
doi = {10.1038/s41467-025-64852-7},
year = {2025},
date = {2025-12-15},
urldate = {2025-12-15},
journal = {Nature Communications},
volume = {16},
number = {11240},
abstract = {Oriented attachment is a critical, yet poorly understood, crystal growth pathway based on the self-assembly of nanocrystals. During oriented attachment, solvent-separated particles align and coalesce through forces that enable precise rotation and translation. While prior studies emphasized intragap forces driving crystallographic alignment, the forces enabling uniform stacking and superlattice formation remain unclear. Here, we demonstrate how macroscopic gibbsite mesocrystals emerge from nanoplates guided into staggered positions by directional sliding. Electron microscopy and X-ray scattering reveal the monoclinic superlattice structure, based on nanoplate stacking with a uniform ≈50° stagger along the gibbsite [010] direction. In situ liquid-cell TEM captures preferential sliding along the gibbsite [010] direction, decelerating with increasing particle overlap. Molecular dynamics simulations reveal that this staggered arrangement corresponds to a global free-energy minimum, rather than full alignment. The simulations also confirm that sliding along the [010] direction is energetically favored and provide insight into the role of interfacial water in achieving long-range ordered assemblies. These insights highlight the energy landscape’s role in oriented attachment, with implications for material synthesis and hierarchical structures in nature.},
note = {Oriented growth is an important pathway for crystal growth. Here, the authors show that gibbsite nanoplates form mesocrystals through directed sliding and staggered stacking, as demonstrated by in situ microscopy and molecular simulations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oriented attachment is a critical, yet poorly understood, crystal growth pathway based on the self-assembly of nanocrystals. During oriented attachment, solvent-separated particles align and coalesce through forces that enable precise rotation and translation. While prior studies emphasized intragap forces driving crystallographic alignment, the forces enabling uniform stacking and superlattice formation remain unclear. Here, we demonstrate how macroscopic gibbsite mesocrystals emerge from nanoplates guided into staggered positions by directional sliding. Electron microscopy and X-ray scattering reveal the monoclinic superlattice structure, based on nanoplate stacking with a uniform ≈50° stagger along the gibbsite [010] direction. In situ liquid-cell TEM captures preferential sliding along the gibbsite [010] direction, decelerating with increasing particle overlap. Molecular dynamics simulations reveal that this staggered arrangement corresponds to a global free-energy minimum, rather than full alignment. The simulations also confirm that sliding along the [010] direction is energetically favored and provide insight into the role of interfacial water in achieving long-range ordered assemblies. These insights highlight the energy landscape’s role in oriented attachment, with implications for material synthesis and hierarchical structures in nature. |
| 273. | | Xiaohua Qiao, Ruifeng Qi, Junqi Liu, Qingsong Huang Dynamic magnetic fields transform pivoting H2SiO3 particles to hollow microspheres with nanofibrous interior In: Nature Communications, vol. 17, no. 509, 2025, (Static magnetic fields can only interact marginally with non-magnetic materials. Here, the authors show that dynamic magnetic flux can rotate non-magnetic particles and transform them into microstructures with nanoscale fibrous textures.). @article{nokey,
title = {Dynamic magnetic fields transform pivoting H2SiO3 particles to hollow microspheres with nanofibrous interior},
author = {Xiaohua Qiao and Ruifeng Qi and Junqi Liu and Qingsong Huang },
url = {https://www.nature.com/articles/s41467-025-67205-6.pdf},
doi = {10.1038/s41467-025-67205-6},
year = {2025},
date = {2025-12-11},
urldate = {2025-12-11},
journal = {Nature Communications},
volume = {17},
number = {509},
abstract = {Reorganizing the internal structures of hollow microspheres after shell sealing, without damaging the shell, remains a major challenge. Here, we show a dynamic magnetic field fluxes stirring (DMFFS) technology that can transform H2SiO3 particle to a sealed SiO2 hollow microsphere with fibrous interior within 30 s. The fibrous structure is composed of SiO2 micro-nano fibers (MNF). As dynamic magnetic field drives the SiO2 particles to rotate, the rotated SiO2 particles become magnetized along the rotation axis according to the Barnett effect. Magnetized SiO2 interacts with magnetic field to form fibers. This MNF-structured microsphere exhibits low density (0.1 g cm–3), high transparency (ca. 85.6%), superior thermal stability (1200 °C) and thermal insulation performance. Fluorescent additives can be uniformly incorporated into the MNF-structured microspheres, enabling distinctive and reproducible fluorescent functionality. In addition, a sealed SiO2 hollow microsphere with nanofibrous interior can be obtained by extending the stirring time or increasing the magnetic field strength. The DMFFS process provides an alternative route to rapidly produce mass SiO2 microspheres containing SiO2 micro/nano-sized fibers. Furthermore, our findings have provided an insight for manipulating non-magnetic materials.},
note = {Static magnetic fields can only interact marginally with non-magnetic materials. Here, the authors show that dynamic magnetic flux can rotate non-magnetic particles and transform them into microstructures with nanoscale fibrous textures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Reorganizing the internal structures of hollow microspheres after shell sealing, without damaging the shell, remains a major challenge. Here, we show a dynamic magnetic field fluxes stirring (DMFFS) technology that can transform H2SiO3 particle to a sealed SiO2 hollow microsphere with fibrous interior within 30 s. The fibrous structure is composed of SiO2 micro-nano fibers (MNF). As dynamic magnetic field drives the SiO2 particles to rotate, the rotated SiO2 particles become magnetized along the rotation axis according to the Barnett effect. Magnetized SiO2 interacts with magnetic field to form fibers. This MNF-structured microsphere exhibits low density (0.1 g cm–3), high transparency (ca. 85.6%), superior thermal stability (1200 °C) and thermal insulation performance. Fluorescent additives can be uniformly incorporated into the MNF-structured microspheres, enabling distinctive and reproducible fluorescent functionality. In addition, a sealed SiO2 hollow microsphere with nanofibrous interior can be obtained by extending the stirring time or increasing the magnetic field strength. The DMFFS process provides an alternative route to rapidly produce mass SiO2 microspheres containing SiO2 micro/nano-sized fibers. Furthermore, our findings have provided an insight for manipulating non-magnetic materials. |
| 272. | | Xiao Wei, Mingliang Li, Xinyue Chang, Ziqi Song, Ping Duan, Chuancheng Jia, Xuefeng Guo Effect of hydrogen migration on the electronic structure of phenol derivatives observed by single-molecule conductance In: Nature Communications, vol. 16, no. 10864 , 2025, (Regulation of the electronic structure in aromatic systems has a significant influence on their chemical reactivity. Here, the authors employ single-molecule junctions to precisely tune the position of a hydrogen atom through the electric field.). @article{nokey,
title = {Effect of hydrogen migration on the electronic structure of phenol derivatives observed by single-molecule conductance},
author = {Xiao Wei and Mingliang Li and Xinyue Chang and Ziqi Song and Ping Duan and Chuancheng Jia and Xuefeng Guo },
url = {https://www.nature.com/articles/s41467-025-65886-7.pdf},
doi = {10.1038/s41467-025-65886-7},
year = {2025},
date = {2025-12-03},
journal = {Nature Communications},
volume = {16},
number = {10864 },
abstract = {The regulation of the electronic structure of an aromatic system significantly influences its chemical reactivity, optical behavior, and electrical property. A comprehensive understanding of the impact of hydrogen atom states on the electronic structure of phenol derivatives and the ability to regulate these effects is crucial for the design and development of functional materials. Here, we employ a single-molecule junction technology to simultaneously monitor and regulate a single hydrogen atom’s state in phenol derivatives. We show that the combined effects of the applied electric field and the acid-base environment in molecular solutions enable hydrogen bonding interactions to precisely control the movement of the hydrogen atom on phenol derivatives, drawing it closer to or farther away from the oxygen atom depending on the external stimuli. Complementary theoretical calculations reveal how these state changes alter electron distribution and charge transport pathways, consequently impacting molecular conductance. This study provides insights for developing controllable molecular devices.},
note = {Regulation of the electronic structure in aromatic systems has a significant influence on their chemical reactivity. Here, the authors employ single-molecule junctions to precisely tune the position of a hydrogen atom through the electric field.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The regulation of the electronic structure of an aromatic system significantly influences its chemical reactivity, optical behavior, and electrical property. A comprehensive understanding of the impact of hydrogen atom states on the electronic structure of phenol derivatives and the ability to regulate these effects is crucial for the design and development of functional materials. Here, we employ a single-molecule junction technology to simultaneously monitor and regulate a single hydrogen atom’s state in phenol derivatives. We show that the combined effects of the applied electric field and the acid-base environment in molecular solutions enable hydrogen bonding interactions to precisely control the movement of the hydrogen atom on phenol derivatives, drawing it closer to or farther away from the oxygen atom depending on the external stimuli. Complementary theoretical calculations reveal how these state changes alter electron distribution and charge transport pathways, consequently impacting molecular conductance. This study provides insights for developing controllable molecular devices. |
| 271. | | Felix Stete, Matias Bargheer, Wouter Koopman Capacitive photocharging of gold nanorods In: Nature Communications, vol. 17, no. 139, 2025, (Light can charge metal nanoparticles. Here, the authors present a method that allows the charging to be observed in situ, making it possible to model the process quantitatively as nanorods that behave like photocharged nanocapacitors.). @article{nokey,
title = {Capacitive photocharging of gold nanorods},
author = {Felix Stete and Matias Bargheer and Wouter Koopman },
url = {https://www.nature.com/articles/s41467-025-67130-8.pdf},
doi = {10.1038/s41467-025-67130-8},
year = {2025},
date = {2025-12-03},
urldate = {2025-12-03},
journal = {Nature Communications},
volume = {17},
number = {139},
abstract = {Light can charge plasmonic nanoparticles by photoredox reactions, significantly modifying their optical and chemical properties. However, the charging process has been challenging to track experimentally, severely hindering its thorough evaluation. In this study, we investigate the charging of gold nanorods during a light-induced reaction in situ, utilizing the sensitivity of the rods’ longitudinal localized surface plasmon resonance to charge accumulation. Describing the particles as nanocapacitors, we present a model to quantify the number of charges on the particles and their connection to the illumination intensity. We find that the Fermi level, together with all other energy bands, is raised because of the repulsive potential of the additional charges. Experimental observations of the dependence on the solvent, the particle size, and ligand type further corroborate the proposed capacitor model. The results presented in this study lay the groundwork for the rational engineering of dynamic charge accumulation during plasmon-driven photoreactions.},
note = {Light can charge metal nanoparticles. Here, the authors present a method that allows the charging to be observed in situ, making it possible to model the process quantitatively as nanorods that behave like photocharged nanocapacitors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Light can charge plasmonic nanoparticles by photoredox reactions, significantly modifying their optical and chemical properties. However, the charging process has been challenging to track experimentally, severely hindering its thorough evaluation. In this study, we investigate the charging of gold nanorods during a light-induced reaction in situ, utilizing the sensitivity of the rods’ longitudinal localized surface plasmon resonance to charge accumulation. Describing the particles as nanocapacitors, we present a model to quantify the number of charges on the particles and their connection to the illumination intensity. We find that the Fermi level, together with all other energy bands, is raised because of the repulsive potential of the additional charges. Experimental observations of the dependence on the solvent, the particle size, and ligand type further corroborate the proposed capacitor model. The results presented in this study lay the groundwork for the rational engineering of dynamic charge accumulation during plasmon-driven photoreactions. |
| 270. | | Feng Gao, Chunlei Yang, Yuanhao Lyu, Luhao Zhang, Peng Cheng, Lan Chen, En-Ge Wang, Svetlana Klyatskaya, Cui Zhang, Mario Ruben, Sheng Meng, Kehui Wu, Yi-Qi Zhang Anomalous rotational-symmetry breaking in proton arrangement of surface-confined cyclic hydrogen bonds revealed by atomic force spectroscopy In: Nature Communications, vol. 16, no. 153, 2025, (Detecting proton arrangements and quantum behavior at the atomic level is a challenge. Here, the authors use atomic force spectroscopy to reveal symmetry breaking in the proton arrangement in cyclic hydrogen bonds caused by nuclear quantum effects.). @article{nokey,
title = {Anomalous rotational-symmetry breaking in proton arrangement of surface-confined cyclic hydrogen bonds revealed by atomic force spectroscopy},
author = {Feng Gao and Chunlei Yang and Yuanhao Lyu and Luhao Zhang and Peng Cheng and Lan Chen and En-Ge Wang and Svetlana Klyatskaya and Cui Zhang and Mario Ruben and Sheng Meng and Kehui Wu and Yi-Qi Zhang },
url = {https://www.nature.com/articles/s41467-025-66848-9.pdf},
doi = {10.1038/s41467-025-66848-9},
year = {2025},
date = {2025-12-02},
urldate = {2025-12-02},
journal = {Nature Communications},
volume = {16},
number = {153},
abstract = {In hydrogen-bonded materials and biosystems, microscopic proton ordering, which is strongly influenced by the quantum nature of protons, underpins diverse macroscopic phenomena, including phase transitions, chemical reactions, biomolecular processes and collective proton transfer. Yet resolving proton arrangements and characterizing their quantum behavior at the atomic scale remain challenging due to the small size of protons and the lack of effective approaches. Here, we exploit bond-resolved atomic force microscopy and spectroscopy (BR-AFM/AFS) to probe signatures of proton ordering in surface-confined benzimidazole (BI) assemblies. By performing BR-AFS along the apparent H-bond between proton donor and acceptor nitrogen atoms, we extract information consistent with H-bonding directionality and signatures compatible with quantum proton delocalization. We observe anomalous rotational-symmetry breaking in the proton order of the cyclic hexamers, arising from the coexistence of both localized and quantum-delocalized protons. The chirality of a single hexamer can be reversibly switched by altering its adsorption registry combined with collective transfer of six protons. Path-integral molecular dynamics calculations unravel that nuclear quantum effects promote proton delocalization and concomitant reduction of donor-acceptor distance. Our findings enable atomic-level detection of complex proton orders, engineering proton-based quantum states, and elucidation of long-range proton transfer mechanisms in molecular architectures.},
note = {Detecting proton arrangements and quantum behavior at the atomic level is a challenge. Here, the authors use atomic force spectroscopy to reveal symmetry breaking in the proton arrangement in cyclic hydrogen bonds caused by nuclear quantum effects.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In hydrogen-bonded materials and biosystems, microscopic proton ordering, which is strongly influenced by the quantum nature of protons, underpins diverse macroscopic phenomena, including phase transitions, chemical reactions, biomolecular processes and collective proton transfer. Yet resolving proton arrangements and characterizing their quantum behavior at the atomic scale remain challenging due to the small size of protons and the lack of effective approaches. Here, we exploit bond-resolved atomic force microscopy and spectroscopy (BR-AFM/AFS) to probe signatures of proton ordering in surface-confined benzimidazole (BI) assemblies. By performing BR-AFS along the apparent H-bond between proton donor and acceptor nitrogen atoms, we extract information consistent with H-bonding directionality and signatures compatible with quantum proton delocalization. We observe anomalous rotational-symmetry breaking in the proton order of the cyclic hexamers, arising from the coexistence of both localized and quantum-delocalized protons. The chirality of a single hexamer can be reversibly switched by altering its adsorption registry combined with collective transfer of six protons. Path-integral molecular dynamics calculations unravel that nuclear quantum effects promote proton delocalization and concomitant reduction of donor-acceptor distance. Our findings enable atomic-level detection of complex proton orders, engineering proton-based quantum states, and elucidation of long-range proton transfer mechanisms in molecular architectures. |
| 269. | | Li Tang, Wei Zhang, Qikai Han, Bin Wang, Meng Zhou, Shuxin Wang Sub-ångström bond length tuning enhances photoluminescence quantum yield in copper nanoclusters In: Nature Communications, vol. 16, no. 10715, 2025, (Metal nanoclusters exhibit size-dependent photoluminescence, but a quantitative link between structure and emission is rare. Here, the authors tune Cu-Cu bond distances in Cu6(SR)6 clusters and show a direct exponential relationship to quantum yield.). @article{nokey,
title = {Sub-ångström bond length tuning enhances photoluminescence quantum yield in copper nanoclusters},
author = {Li Tang and Wei Zhang and Qikai Han and Bin Wang and Meng Zhou and Shuxin Wang },
url = {https://www.nature.com/articles/s41467-025-65739-3.pdf},
doi = {10.1038/s41467-025-65739-3},
year = {2025},
date = {2025-11-28},
journal = {Nature Communications},
volume = {16},
number = {10715},
abstract = {Understanding how atomic-scale structure dictates light emission in metal nanoclusters is central to designing efficient luminophores. Despite decades of intensive investigation into their photoluminescence, a clear quantitative link between metal-metal bonding and emission efficiency is still lacking. Here we show that quantitatively modulating Cu-Cu bond distances during crystallization of Cu6(SR)6 nanoclusters enables a direct correlation between structure and emission performance. By synthesizing a series of Cu6(SR)6 nanoclusters with quantitatively modulated Cu-Cu bond lengths, we reveal an exponential relationship between bond distance and photoluminescence quantum yield (PLQY), and a linear correlation with emission energy. Density functional theory (DFT) calculations and ultrafast spectroscopy demonstrate that the enhanced PLQY arises from reduced HOMO-LUMO overlap induced by extended Cu-Cu distances, which promotes greater orbital localization. Simultaneously, the associated widening of the electronic gap suppresses non-radiative decay via the energy-gap law, further contributing to the increase in PLQY. This work establishes a quantitative relationship between Cu-Cu bond distance and quantum yield in Cu clusters, providing a general design framework for achieving high-efficiency emitters through quantitative bond-length engineering.},
note = {Metal nanoclusters exhibit size-dependent photoluminescence, but a quantitative link between structure and emission is rare. Here, the authors tune Cu-Cu bond distances in Cu6(SR)6 clusters and show a direct exponential relationship to quantum yield.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Understanding how atomic-scale structure dictates light emission in metal nanoclusters is central to designing efficient luminophores. Despite decades of intensive investigation into their photoluminescence, a clear quantitative link between metal-metal bonding and emission efficiency is still lacking. Here we show that quantitatively modulating Cu-Cu bond distances during crystallization of Cu6(SR)6 nanoclusters enables a direct correlation between structure and emission performance. By synthesizing a series of Cu6(SR)6 nanoclusters with quantitatively modulated Cu-Cu bond lengths, we reveal an exponential relationship between bond distance and photoluminescence quantum yield (PLQY), and a linear correlation with emission energy. Density functional theory (DFT) calculations and ultrafast spectroscopy demonstrate that the enhanced PLQY arises from reduced HOMO-LUMO overlap induced by extended Cu-Cu distances, which promotes greater orbital localization. Simultaneously, the associated widening of the electronic gap suppresses non-radiative decay via the energy-gap law, further contributing to the increase in PLQY. This work establishes a quantitative relationship between Cu-Cu bond distance and quantum yield in Cu clusters, providing a general design framework for achieving high-efficiency emitters through quantitative bond-length engineering. |
| 268. | | Phillip Seiler, Anthony J. R. Payne, Neubi F. Xavier Jr, Louie Slocombe, Marco Sacchi, Anton Tamtögl Understanding water behaviour on 2D material interfaces through single-molecule motion on h-BN and graphene In: Nature Communications, vol. 16, no. 10465, 2025, (Water interactions with 2D materials shape sensing and fluid control. Here, the authors show that water diffuses on hexagonal boron nitride with distinct rotational dynamics and lower friction than on graphene, revealing substrate effects on interfacial water.). @article{nokey,
title = {Understanding water behaviour on 2D material interfaces through single-molecule motion on h-BN and graphene},
author = {Phillip Seiler and Anthony J. R. Payne and Neubi F. Xavier Jr and Louie Slocombe and Marco Sacchi and Anton Tamtögl },
url = {https://www.nature.com/articles/s41467-025-65452-1.pdf},
doi = {10.1038/s41467-025-65452-1},
year = {2025},
date = {2025-11-25},
journal = {Nature Communications},
volume = {16},
number = {10465},
abstract = {Understanding water behaviour on 2D materials is crucial for applications in sensing, microfluidics, and tribology. While graphene-water interactions are well studied, water on hexagonal boron nitride (h-BN) remains largely unexplored. Despite its structural similarity to graphene, h-BN possesses polar B-N bonds that give rise to distinct electronic and chemical properties. Most previous studies have also focused on multilayer water, leaving single-molecule dynamics poorly understood. Here we show how individual water molecules diffuse on h-BN compared to graphene using helium spin-echo spectroscopy and ab initio calculations. On h-BN/Ni, water exhibits coupled rotational-translational motion, in contrast to the discrete hopping observed on graphene. Water molecules rotate freely around their centre of mass, and although binding energies are similar on both materials, the activation energy for water dynamics on h-BN is 2.5 times lower than on graphene. These dynamics, which classical models fail to capture, highlight the fundamentally different nature of water transport on polar 2D surfaces. We further demonstrate that the supporting substrate strongly influences water friction, with h-BN/Ni showing markedly lower friction than graphene/Ni, opposite to the behaviour of free-standing layers. These findings challenge assumptions and offer insights for designing microfluidic devices requiring precise control of water mobility.},
note = {Water interactions with 2D materials shape sensing and fluid control. Here, the authors show that water diffuses on hexagonal boron nitride with distinct rotational dynamics and lower friction than on graphene, revealing substrate effects on interfacial water.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Understanding water behaviour on 2D materials is crucial for applications in sensing, microfluidics, and tribology. While graphene-water interactions are well studied, water on hexagonal boron nitride (h-BN) remains largely unexplored. Despite its structural similarity to graphene, h-BN possesses polar B-N bonds that give rise to distinct electronic and chemical properties. Most previous studies have also focused on multilayer water, leaving single-molecule dynamics poorly understood. Here we show how individual water molecules diffuse on h-BN compared to graphene using helium spin-echo spectroscopy and ab initio calculations. On h-BN/Ni, water exhibits coupled rotational-translational motion, in contrast to the discrete hopping observed on graphene. Water molecules rotate freely around their centre of mass, and although binding energies are similar on both materials, the activation energy for water dynamics on h-BN is 2.5 times lower than on graphene. These dynamics, which classical models fail to capture, highlight the fundamentally different nature of water transport on polar 2D surfaces. We further demonstrate that the supporting substrate strongly influences water friction, with h-BN/Ni showing markedly lower friction than graphene/Ni, opposite to the behaviour of free-standing layers. These findings challenge assumptions and offer insights for designing microfluidic devices requiring precise control of water mobility. |
| 267. | | Aleksandra Borkenhagen, Kamil Sokołowski, Paweł W. Majewski, Iwona Justyniak, Paula Roś, Anna M. Cieślak, Janusz Lewiński Chemical weathering of molecular single crystals to monoliths of quantum dots In: Nature Communications, vol. 16, no. 10254, 2025, (Artificial systems that mechanistically parallel geological nanocrystal formation remain rare. Here, the authors show the solid-state synthesis of semiconductor quantum dots from a submillimeter-sized molecular single crystal under ambient conditions, mimicking natural weathering.). @article{nokey,
title = {Chemical weathering of molecular single crystals to monoliths of quantum dots},
author = {Aleksandra Borkenhagen and Kamil Sokołowski and Paweł W. Majewski and Iwona Justyniak and Paula Roś and Anna M. Cieślak and Janusz Lewiński },
url = {https://www.nature.com/articles/s41467-025-65113-3.pdf},
doi = {10.1038/s41467-025-65113-3},
year = {2025},
date = {2025-11-21},
journal = {Nature Communications},
volume = {16},
number = {10254},
abstract = {In nature, nanocrystals form primarily through geological processes, such as chemical weathering of rocks and minerals. However, artificial systems that mechanistically parallel geological nanocrystal formation, particularly in the solid state, replicating the hydrolytic conditions of geological processes remain rare. Here, we report an all-solid-state strategy for the synthesis of quantum-sized semiconductor nanostructured materials under ambient conditions. The approach relies on a hydrolysable organozinc precursor, consisting of [RZn(amidate)]n-type molecular clusters featuring reactive Zn-C and Zn-N bonds, confined within a single-crystal lattice prone to water-mediated hydrolytic processes. The initial millimetre-sized precursor crystals, when exposed to humid air, undergo hydrolytic transformations. Within hours these transformations lead to the formation of ZnO quantum dots at room temperature, reminiscent of natural chemical weathering. The crystalline network of the precursor system provides a specific environment for the formation of zinc hydroxide/oxide species, followed by nucleation and growth of quantum dots within an emerging hydrogen-bonded host organic matrix composed from the hydrolysable amidate ligands. This organic matrix, in turn, can be removed from the system under reduced pressure, yielding a nanocrystalline mesoporous scaffold consisting of quantum-sized crystals. This organometallic synthetic system provides insights into solid-state nanostructured materials formation under mild conditions and offers a pathway to sustainable synthesis of functional materials.},
note = {Artificial systems that mechanistically parallel geological nanocrystal formation remain rare. Here, the authors show the solid-state synthesis of semiconductor quantum dots from a submillimeter-sized molecular single crystal under ambient conditions, mimicking natural weathering.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In nature, nanocrystals form primarily through geological processes, such as chemical weathering of rocks and minerals. However, artificial systems that mechanistically parallel geological nanocrystal formation, particularly in the solid state, replicating the hydrolytic conditions of geological processes remain rare. Here, we report an all-solid-state strategy for the synthesis of quantum-sized semiconductor nanostructured materials under ambient conditions. The approach relies on a hydrolysable organozinc precursor, consisting of [RZn(amidate)]n-type molecular clusters featuring reactive Zn-C and Zn-N bonds, confined within a single-crystal lattice prone to water-mediated hydrolytic processes. The initial millimetre-sized precursor crystals, when exposed to humid air, undergo hydrolytic transformations. Within hours these transformations lead to the formation of ZnO quantum dots at room temperature, reminiscent of natural chemical weathering. The crystalline network of the precursor system provides a specific environment for the formation of zinc hydroxide/oxide species, followed by nucleation and growth of quantum dots within an emerging hydrogen-bonded host organic matrix composed from the hydrolysable amidate ligands. This organic matrix, in turn, can be removed from the system under reduced pressure, yielding a nanocrystalline mesoporous scaffold consisting of quantum-sized crystals. This organometallic synthetic system provides insights into solid-state nanostructured materials formation under mild conditions and offers a pathway to sustainable synthesis of functional materials. |
| 266. | | Weilin Zheng, Jiangkun Chen, Yang Guo, Xin Zhang, Fengjun Chun, Yaxin Cao, Xiaoping Gao, Hao Suo, Xiaohe Wei, Zhifeng Xing, Yanze Wang, Feng Wang Pb deficiency enables the synthesis of CsPbI3 nanosheets with tunable thickness down to two octahedral layers In: Nature Communications, vol. 16, no. 10078, 2025, (CsPbI3 nanolayers of two-octahedral layer thickness are required for full-color emission in mono-halide perovskites, but their synthesis is complicated by the high reactivity of I-containing crystallites. Here, the authors devise Pb deficiency to retard crystal growth and thus limit growth to two octahedral layers.). @article{nokey,
title = {Pb deficiency enables the synthesis of CsPbI3 nanosheets with tunable thickness down to two octahedral layers},
author = {Weilin Zheng and Jiangkun Chen and Yang Guo and Xin Zhang and Fengjun Chun and Yaxin Cao and Xiaoping Gao and Hao Suo and Xiaohe Wei and Zhifeng Xing and Yanze Wang and Feng Wang },
url = {https://www.nature.com/articles/s41467-025-65050-1.pdf},
doi = {10.1038/s41467-025-65050-1},
year = {2025},
date = {2025-11-18},
journal = {Nature Communications},
volume = {16},
number = {10078},
abstract = {CsPbX3 (X = Cl, Br, I) nanosheets have emerged as a promising platform for tuning quantum confinement effects and achieving efficient optoelectronic devices. However, synthesizing uniform CsPbX3 nanosheets with monolayer-level thicknesses remains a challenge due to the fast reaction kinetics. Herein, we develop a synthetic strategy that leverages Pb-deficient precursors to control reaction kinetics. Our experimental investigations indicate that Pb deficiency in the precursors decelerates the crystal growth, offering enhanced dimensional control in CsPbX3 systems. This approach enables the synthesis of uniform CsPbI3 nanosheets with tunable thickness down to two octahedral layers, which exhibit a narrowband emission at 563 nm and high stability against spectral drift. This method is readily extended to CsPbBr3 and CsPbCl3 systems for precise size and shape modulation, thus affording full-color emission tuning within mono-halide perovskites. These findings establish a versatile synthetic strategy poised to advance the design and application of CsPbX3 nanomaterials and other colloidal nanocrystals.},
note = {CsPbI3 nanolayers of two-octahedral layer thickness are required for full-color emission in mono-halide perovskites, but their synthesis is complicated by the high reactivity of I-containing crystallites. Here, the authors devise Pb deficiency to retard crystal growth and thus limit growth to two octahedral layers.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
CsPbX3 (X = Cl, Br, I) nanosheets have emerged as a promising platform for tuning quantum confinement effects and achieving efficient optoelectronic devices. However, synthesizing uniform CsPbX3 nanosheets with monolayer-level thicknesses remains a challenge due to the fast reaction kinetics. Herein, we develop a synthetic strategy that leverages Pb-deficient precursors to control reaction kinetics. Our experimental investigations indicate that Pb deficiency in the precursors decelerates the crystal growth, offering enhanced dimensional control in CsPbX3 systems. This approach enables the synthesis of uniform CsPbI3 nanosheets with tunable thickness down to two octahedral layers, which exhibit a narrowband emission at 563 nm and high stability against spectral drift. This method is readily extended to CsPbBr3 and CsPbCl3 systems for precise size and shape modulation, thus affording full-color emission tuning within mono-halide perovskites. These findings establish a versatile synthetic strategy poised to advance the design and application of CsPbX3 nanomaterials and other colloidal nanocrystals. |
| 265. | | Peiyao Pan, Weinan Dong, Wentao Huang, Xue Bai, Zhennan Wu, Xi Kang, Manzhou Zhu Regulation of the photophysical dynamics of metal nanoclusters by manipulating single-point defects In: Nature Communications, vol. 16, no. 10065, 2025, (The properties of atomically precise nanoclusters depend on their structural composition, but controlling them is challenging. Here, the authors demonstrate the manipulation of single-point defects, converting Au21 into Au22 and thereby regulating the photophysical dynamics.). @article{nokey,
title = {Regulation of the photophysical dynamics of metal nanoclusters by manipulating single-point defects},
author = {Peiyao Pan and Weinan Dong and Wentao Huang and Xue Bai and Zhennan Wu and Xi Kang and Manzhou Zhu },
url = {https://www.nature.com/articles/s41467-025-65024-3.pdf},
doi = {10.1038/s41467-025-65024-3},
year = {2025},
date = {2025-11-17},
journal = {Nature Communications},
volume = {16},
number = {10065},
abstract = {Metal nanoclusters have served as an emerging class of programmable nanomaterials with customized structures. However, it remains highly challenging to achieve the single-atom regulation of metal nanoclusters without altering their structural frameworks. Here, we achieve the single-point defects manipulation based upon a cluster pair of Au21 and Au22 by meticulously complementing the surface defects of the former nanocluster with an additional single-Au complex. The two nanoclusters exhibited identical geometric structures, but their pronounced quantum-confinement effects resulted in different electronic properties, evident in their distinct optical absorption and emission characteristics. Temperature-dependent steady-state photoluminescence spectra and femtosecond transient absorption spectra showed that the manipulation of a single-point defect in Au22 inhibited non-radiative decay pathways, reduced electron loss at higher energy levels, and accelerated intersystem crossing, which ultimately enhanced its emission intensity. Overall, the Au21 and Au22 cluster system in this study provides a cluster platform with controllable surface single-point defects, enabling the regulation of the photophysical dynamics at the atomic level.},
note = {The properties of atomically precise nanoclusters depend on their structural composition, but controlling them is challenging. Here, the authors demonstrate the manipulation of single-point defects, converting Au21 into Au22 and thereby regulating the photophysical dynamics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Metal nanoclusters have served as an emerging class of programmable nanomaterials with customized structures. However, it remains highly challenging to achieve the single-atom regulation of metal nanoclusters without altering their structural frameworks. Here, we achieve the single-point defects manipulation based upon a cluster pair of Au21 and Au22 by meticulously complementing the surface defects of the former nanocluster with an additional single-Au complex. The two nanoclusters exhibited identical geometric structures, but their pronounced quantum-confinement effects resulted in different electronic properties, evident in their distinct optical absorption and emission characteristics. Temperature-dependent steady-state photoluminescence spectra and femtosecond transient absorption spectra showed that the manipulation of a single-point defect in Au22 inhibited non-radiative decay pathways, reduced electron loss at higher energy levels, and accelerated intersystem crossing, which ultimately enhanced its emission intensity. Overall, the Au21 and Au22 cluster system in this study provides a cluster platform with controllable surface single-point defects, enabling the regulation of the photophysical dynamics at the atomic level. |
| 264. | | Fei Chen, Junkang Shan, Xi Wang, Jingning Li, Fei Jia, Fan Wang, Zihao Yang, Kai Liu, Ru-Wen Peng, Mu Wang Electrodeposition of magnetic nanonetworks featuring triangular motifs and parallel ridges on a macroscopic scale In: Nature Communications, vol. 16, no. 9965, 2025, (Self-organized growth often suffers from a lack of long-range order in preparing large-area nanostructures. Here, the authors develop a direct electrochemical growth technique for preparing ordered magnetic networks without the use of templates.). @article{nokey,
title = {Electrodeposition of magnetic nanonetworks featuring triangular motifs and parallel ridges on a macroscopic scale},
author = {Fei Chen and Junkang Shan and Xi Wang and Jingning Li and Fei Jia and Fan Wang and Zihao Yang and Kai Liu and Ru-Wen Peng and Mu Wang },
url = {https://www.nature.com/articles/s41467-025-65589-z.pdf},
doi = {10.1038/s41467-025-65589-z},
year = {2025},
date = {2025-11-14},
journal = {Nature Communications},
volume = {16},
number = {9965},
abstract = {Ordered nanostructured magnetic networks offer a versatile platform for studying magnetic frustration, reconfigurable magnonics, and neuromorphic computing. Their fabrication, however, often relies on complicated and expensive nanolithography. Here, we demonstrate an electrochemical strategy for growing large-area magnetic networks composed of self-organized triangular motifs separated by periodic parallel ridges. The ridges arise deterministically from voltage-controlled electrodeposition, while the regions between them undergo self-organization: cobalt nucleates, bifurcates, and assembles into either solid or hollow triangular units depending on the growth conditions. The global network morphology is dictated by the waveform of the applied voltage. These triangular structures host diverse magnetic states, including vortex-like and macro-spin-like configurations, and exhibit nonlinear responses to external magnetic fields. Our approach combines macroscopically controlled, deterministic growth and microscopic self-organization, providing a scalable pathway for fabricating complex nanostructures. The resulting magnetic networks show potential as physical reservoirs for neuromorphic computing.},
note = {Self-organized growth often suffers from a lack of long-range order in preparing large-area nanostructures. Here, the authors develop a direct electrochemical growth technique for preparing ordered magnetic networks without the use of templates.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ordered nanostructured magnetic networks offer a versatile platform for studying magnetic frustration, reconfigurable magnonics, and neuromorphic computing. Their fabrication, however, often relies on complicated and expensive nanolithography. Here, we demonstrate an electrochemical strategy for growing large-area magnetic networks composed of self-organized triangular motifs separated by periodic parallel ridges. The ridges arise deterministically from voltage-controlled electrodeposition, while the regions between them undergo self-organization: cobalt nucleates, bifurcates, and assembles into either solid or hollow triangular units depending on the growth conditions. The global network morphology is dictated by the waveform of the applied voltage. These triangular structures host diverse magnetic states, including vortex-like and macro-spin-like configurations, and exhibit nonlinear responses to external magnetic fields. Our approach combines macroscopically controlled, deterministic growth and microscopic self-organization, providing a scalable pathway for fabricating complex nanostructures. The resulting magnetic networks show potential as physical reservoirs for neuromorphic computing. |
| 263. | | Eunyoung Park, Chulho Jung, Junha Hwang, Jaeyong Shin, Sung Yun Lee, Heemin Lee, Seung-Phil Heo, Daewoong Nam, Sangsoo Kim, Min Seok Kim, Kyung Sook Kim, In Tae Eom, Yungok Ihm, Do Young Noh, Changyong Song Surface-plasmon control of ultrafast energy-relaxation modes in photoexcited Au nanorods probed by time-resolved single-particle X-ray imaging 2025, (Ultrafast lasers can drive materials into states beyond equilibrium. Here, the authors use single-pulse X-ray imaging to show that surface plasmons channel energy relaxation into distinct pathways, leading to different shape deformations.). @bachelorthesis{nokey,
title = {Surface-plasmon control of ultrafast energy-relaxation modes in photoexcited Au nanorods probed by time-resolved single-particle X-ray imaging},
author = {Eunyoung Park and Chulho Jung and Junha Hwang and Jaeyong Shin and Sung Yun Lee and Heemin Lee and Seung-Phil Heo and Daewoong Nam and Sangsoo Kim and Min Seok Kim and Kyung Sook Kim and In Tae Eom and Yungok Ihm and Do Young Noh and Changyong Song },
url = {https://www.nature.com/articles/s41467-025-64853-6.pdf},
doi = {10.1038/s41467-025-64853-6},
year = {2025},
date = {2025-11-10},
journal = {Nature Communications},
volume = {16},
number = {9876},
abstract = {Ultrafast laser excitation can drive materials into exotic states beyond thermodynamic limits, offering alternative ways to control how matter stores and releases energy. Yet, whether light can actively steer energy-relaxation pathways during structural transitions remains unclear due to the lack of direct experimental evidence. Here we show, using single-pulse time-resolved X-ray imaging of gold nanorods, that photoinduced localized surface plasmons control ultrafast energy relaxation into distinct deformation modes, transverse or longitudinal deformation modes, each accompanied by characteristic plasmon-induced oscillatory distortions depending on the laser fluence. Numerical simulations further confirm that localized surface plasmons dictate ultrafast energy relaxation process from photoexcited hot electrons to anharmonic nanocrystal deformations. Our results provide direct evidence that surface plasmon-mediated interactions enable ultrafast, nanoscale control of materials’ energetics, opening a pathway for tailoring energy-transfer processes with femtosecond laser fields. This approach lays the foundation for customizing nonequilibrium phase dynamics at the nanoscale and provides a route to tailoring energy-transfer processes using femtosecond laser fields.},
note = {Ultrafast lasers can drive materials into states beyond equilibrium. Here, the authors use single-pulse X-ray imaging to show that surface plasmons channel energy relaxation into distinct pathways, leading to different shape deformations.},
keywords = {},
pubstate = {published},
tppubtype = {bachelorthesis}
}
Ultrafast laser excitation can drive materials into exotic states beyond thermodynamic limits, offering alternative ways to control how matter stores and releases energy. Yet, whether light can actively steer energy-relaxation pathways during structural transitions remains unclear due to the lack of direct experimental evidence. Here we show, using single-pulse time-resolved X-ray imaging of gold nanorods, that photoinduced localized surface plasmons control ultrafast energy relaxation into distinct deformation modes, transverse or longitudinal deformation modes, each accompanied by characteristic plasmon-induced oscillatory distortions depending on the laser fluence. Numerical simulations further confirm that localized surface plasmons dictate ultrafast energy relaxation process from photoexcited hot electrons to anharmonic nanocrystal deformations. Our results provide direct evidence that surface plasmon-mediated interactions enable ultrafast, nanoscale control of materials’ energetics, opening a pathway for tailoring energy-transfer processes with femtosecond laser fields. This approach lays the foundation for customizing nonequilibrium phase dynamics at the nanoscale and provides a route to tailoring energy-transfer processes using femtosecond laser fields. |
| 262. | | Ru Lin, Yuyu Xu, Shuo Yang, Shunwei Chen, Zifei Wang, Kang Wang, Zhenhua Gao, Yong Sheng Zhao Regioselective on-surface crystallization of one-dimensional organic barcode-like heterostructures of MOFs with fluorescent stripe patterns In: Nature Communications, vol. 16, no. 9810, 2025, (Heterostructures of segmented semiconductor materials are difficult to produce by sequential epitaxial nucleation. Here, the authors report on the spontaneous formation of 1D heterostructures with numerous fluorescent stripe patterns following an assembly strategy based on Plateau-Rayleigh instability.). @article{nokey,
title = {Regioselective on-surface crystallization of one-dimensional organic barcode-like heterostructures of MOFs with fluorescent stripe patterns},
author = {Ru Lin and Yuyu Xu and Shuo Yang and Shunwei Chen and Zifei Wang and Kang Wang and Zhenhua Gao and Yong Sheng Zhao },
url = {https://www.nature.com/articles/s41467-025-64786-0.pdf},
doi = {10.1038/s41467-025-64786-0},
year = {2025},
date = {2025-11-06},
journal = {Nature Communications},
volume = {16},
number = {9810},
abstract = {Heterostructures, integrating different semiconducting materials in a single structure, are key components for various modern electrical and optoelectronic devices. However, it is difficult to maintain the sequential epitaxial nucleation due to the passivation of crystal seeds, thus resulting in limited spatial segments in heterostructures. Here, we report an interface-recognition assembly strategy assisted by Plateau-Rayleigh instability (PRI) to spontaneously form one-dimensional (1D) heterostructures featuring abundant fluorescent stripe patterns. Guided by 1D metal-organic frameworks (MOFs) templates with exposed electron-donating sites, the de-wetting process promotes the effective regioselective crystallization of solute molecules with electron-withdrawing feature on template surface, thus forming 1D barcode-like heterostructures. The spatial locations and number of the stripes are effectively controlled by PRI and template parameters, while their emission colors are primarily governed by the charge-transfer (CT) interactions between solutes. On this basis, we have achieved full-color and heterogeneous stripe patterns by tuning the CT strengths and controlling the stripe nucleation sequences, thus creating abundant 1D barcode-like heterostructures. Through integrating the photoisomers into stripes, these heterostructures exhibit time-dependent graphical patterns and enable construction of 2D photonic barcodes. Our work offers a supramolecular approach for patterning of well-aligned organic heterostructures and rational design of optoelectronic devices that have potential in anti-counterfeiting applications.},
note = {Heterostructures of segmented semiconductor materials are difficult to produce by sequential epitaxial nucleation. Here, the authors report on the spontaneous formation of 1D heterostructures with numerous fluorescent stripe patterns following an assembly strategy based on Plateau-Rayleigh instability.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Heterostructures, integrating different semiconducting materials in a single structure, are key components for various modern electrical and optoelectronic devices. However, it is difficult to maintain the sequential epitaxial nucleation due to the passivation of crystal seeds, thus resulting in limited spatial segments in heterostructures. Here, we report an interface-recognition assembly strategy assisted by Plateau-Rayleigh instability (PRI) to spontaneously form one-dimensional (1D) heterostructures featuring abundant fluorescent stripe patterns. Guided by 1D metal-organic frameworks (MOFs) templates with exposed electron-donating sites, the de-wetting process promotes the effective regioselective crystallization of solute molecules with electron-withdrawing feature on template surface, thus forming 1D barcode-like heterostructures. The spatial locations and number of the stripes are effectively controlled by PRI and template parameters, while their emission colors are primarily governed by the charge-transfer (CT) interactions between solutes. On this basis, we have achieved full-color and heterogeneous stripe patterns by tuning the CT strengths and controlling the stripe nucleation sequences, thus creating abundant 1D barcode-like heterostructures. Through integrating the photoisomers into stripes, these heterostructures exhibit time-dependent graphical patterns and enable construction of 2D photonic barcodes. Our work offers a supramolecular approach for patterning of well-aligned organic heterostructures and rational design of optoelectronic devices that have potential in anti-counterfeiting applications. |
| 261. | | Yuna Bae, Eun Mi Kim, Jaehun Chun, Zihua Zhu, Trevor H. Moser, Hanlei Zhang, Jaeyoung Heo, Yun Kyung Shin, Hua Zhou, James E. Evans, Emil C. S. Jensen, Kristian S. Mølhave, Kristen A. Fichthorn, James J. De Yoreo, Dongsheng Li Anisotropic surface potentials induced by competitive ion adsorption enable the synthesis of branched cubic Pt mesocrystals In: Nature Communications, vol. 16, no. 9758, 2025, (Nanocrystal assembly is key to creating complex architectures. Here, the authors show that tuning competitive ion adsorption alters anisotropic surface potentials and electrostatic torques, directing facet attachment to enable alternative mesoscale designs.). @article{nokey,
title = {Anisotropic surface potentials induced by competitive ion adsorption enable the synthesis of branched cubic Pt mesocrystals},
author = {Yuna Bae and Eun Mi Kim and Jaehun Chun and Zihua Zhu and Trevor H. Moser and Hanlei Zhang and Jaeyoung Heo and Yun Kyung Shin and Hua Zhou and James E. Evans and Emil C. S. Jensen and Kristian S. Mølhave and Kristen A. Fichthorn and James J. De Yoreo and Dongsheng Li},
url = {https://www.nature.com/articles/s41467-025-64494-9.pdf},
doi = {10.1038/s41467-025-64494-9},
year = {2025},
date = {2025-11-05},
urldate = {2025-11-05},
journal = {Nature Communications},
volume = {16},
number = {9758},
abstract = {Creation of complex nanostructured materials through oriented attachment (OA) requires the manipulation of interparticle forces, including electrostatic repulsion, which depends strongly on surface potentials and can be modified through the effect of solution environment on interfacial chemistry. Here we show that time-dependent anisotropies in surface potential driven by competitive ion adsorption can alter facet-selectivity during OA. This phenomenon enables the synthesis of branched cubic Pt mesocrystals. Initially, Pt nanoparticles attach preferentially at their {100} facets to form a well-defined cubic core. Over time, changes in ion adsorption shift the attachment preference to the {111} facets, promoting branch formation. In both stages, anisotropic surface potentials generate electrostatic torques that align the particles prior to attachment. These findings demonstrate a generalizable strategy for directing the architecture of nanomaterials through time-resolved control of interfacial chemistry during OA, offering new pathways for the design of complex mesoscale structures.},
note = {Nanocrystal assembly is key to creating complex architectures. Here, the authors show that tuning competitive ion adsorption alters anisotropic surface potentials and electrostatic torques, directing facet attachment to enable alternative mesoscale designs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Creation of complex nanostructured materials through oriented attachment (OA) requires the manipulation of interparticle forces, including electrostatic repulsion, which depends strongly on surface potentials and can be modified through the effect of solution environment on interfacial chemistry. Here we show that time-dependent anisotropies in surface potential driven by competitive ion adsorption can alter facet-selectivity during OA. This phenomenon enables the synthesis of branched cubic Pt mesocrystals. Initially, Pt nanoparticles attach preferentially at their {100} facets to form a well-defined cubic core. Over time, changes in ion adsorption shift the attachment preference to the {111} facets, promoting branch formation. In both stages, anisotropic surface potentials generate electrostatic torques that align the particles prior to attachment. These findings demonstrate a generalizable strategy for directing the architecture of nanomaterials through time-resolved control of interfacial chemistry during OA, offering new pathways for the design of complex mesoscale structures. |
| 260. | | Xiaole Ma, Jiaxiang Wang, Jing Li, Yuhan Ren, Di Wu, Zhijuan Su, Jitong Ma, Zhuoqing Yang, Guifu Ding, Faheng Zang Visually encoded mechanoluminescence through hierarchical structuring with microscale patterns and nanoscale features In: Nature Communications, vol. 16, no. 9550, 2025, (Mechanoluminescent films can make forces visible, but brightness and pixelation are limited. Here, the authors develop hierarchical multiscale membranes that enhance emission and enable programmable real-time stress maps with 637 PPI on a single film.). @article{nokey,
title = {Visually encoded mechanoluminescence through hierarchical structuring with microscale patterns and nanoscale features},
author = {Xiaole Ma and Jiaxiang Wang and Jing Li and Yuhan Ren and Di Wu and Zhijuan Su and Jitong Ma and Zhuoqing Yang and Guifu Ding and Faheng Zang },
url = {https://www.nature.com/articles/s41467-025-64558-w.pdf},
doi = {10.1038/s41467-025-64558-w},
year = {2025},
date = {2025-10-29},
journal = {Nature Communications},
volume = {16},
number = {9550},
abstract = {Mechanoluminescence (ML), with its mechano-optical properties, is promising in electronics, photonics, and advanced sensing technologies. However, current thin-film mechanoluminescent devices can hardly provide any pixelated force information attributed to the bottlenecks of their low overall light emission efficiency and poor luminescent contrast. This prevents mechanoluminescent devices from advancing further to full-fledged programmable visual force sensors. Here, we show hierarchical multiscale structures enhanced mechanoluminescent force sensors that leverage the stress-light interaction of multiscale-structured arrays to achieve user-defined pixelated force sensitivity on a uniform ML membrane, delivering a high-resolution output of 637 pixels per inch (PPI). Combining stress localization from the microscale structures and light extraction enhancement from the nanoscale patterns, the ML force-sensing membrane produces a dynamic contrast pattern at 521 nm using a single light-centering material with 366% of light intensity comparing to the bare film. The further demonstration of such multiscale-structured ML in creating force-sensitive counterfeits showcases its potential in developing pixelated and programmable force-light interaction systems.},
note = {Mechanoluminescent films can make forces visible, but brightness and pixelation are limited. Here, the authors develop hierarchical multiscale membranes that enhance emission and enable programmable real-time stress maps with 637 PPI on a single film.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Mechanoluminescence (ML), with its mechano-optical properties, is promising in electronics, photonics, and advanced sensing technologies. However, current thin-film mechanoluminescent devices can hardly provide any pixelated force information attributed to the bottlenecks of their low overall light emission efficiency and poor luminescent contrast. This prevents mechanoluminescent devices from advancing further to full-fledged programmable visual force sensors. Here, we show hierarchical multiscale structures enhanced mechanoluminescent force sensors that leverage the stress-light interaction of multiscale-structured arrays to achieve user-defined pixelated force sensitivity on a uniform ML membrane, delivering a high-resolution output of 637 pixels per inch (PPI). Combining stress localization from the microscale structures and light extraction enhancement from the nanoscale patterns, the ML force-sensing membrane produces a dynamic contrast pattern at 521 nm using a single light-centering material with 366% of light intensity comparing to the bare film. The further demonstration of such multiscale-structured ML in creating force-sensitive counterfeits showcases its potential in developing pixelated and programmable force-light interaction systems. |
| 259. | | Yi Zhou, Qing Zhang, Alvaro Mayoral, Peter R. Spackman, Takashi Matsumoto, Jun Li, Julian D. Gale, Michael W. Anderson, Hiroshi Fukuoka, Shoji Yamanaka, Osamu Terasaki Observation of a guest-free Si46 clathrate-I framework from Ba8-xSi46 upon in situ vacuum heating In: Nature Communications, vol. 16, no. 9238, 2025, (Preparation of a pure Si46 framework in the clathrate-I structure is a challenge. Here, the authors demonstrate the stepwise Ba evacuation process and observe a guest-free Si46 framework from Ba8-xSi46 using atomic-scale STEM imaging during in-situ heating.). @article{nokey,
title = {Observation of a guest-free Si46 clathrate-I framework from Ba8-xSi46 upon in situ vacuum heating},
author = {Yi Zhou and Qing Zhang and Alvaro Mayoral and Peter R. Spackman and Takashi Matsumoto and Jun Li and Julian D. Gale and Michael W. Anderson and Hiroshi Fukuoka and Shoji Yamanaka and Osamu Terasaki },
url = {https://www.nature.com/articles/s41467-025-64277-2.pdf},
doi = {10.1038/s41467-025-64277-2},
year = {2025},
date = {2025-10-17},
journal = {Nature Communications},
volume = {16},
number = {9238},
abstract = {Silicon and its composites are key materials owing to their extensive use in the semiconductor industry. While the diamond-structured form dominates, other allotropes with superior properties at ambient conditions remain of interest. Toward this, Si46 clathrate type-I crystals containing alkali/alkaline-earth metals have been extensively studied, but the experimental observation of a guest-free Si46 structure has been challenging. Using advanced electron microscopy, we show experimental evidence of guest-free clathrate-I Si46 framework from Ba8-xSi46 under in situ heating. We reveal the stepwise Ba evacuation process, starting with loss of Ba1 from the smaller cages to form Ba6Si46, followed by removal of Ba2 in larger cages to reach Si46 that appears in the thin region of the nanocrystal with a thickness around 4-6 nm at 500 °C. Calculations give a quasi-direct bandgap of 1.89 eV and support the preferential evacuation of Ba1. The observation of this guest-free Si46 framework opens up possibilities for applications in high-speed transistors, optoelectronic devices or solar cell technologies.},
note = {Preparation of a pure Si46 framework in the clathrate-I structure is a challenge. Here, the authors demonstrate the stepwise Ba evacuation process and observe a guest-free Si46 framework from Ba8-xSi46 using atomic-scale STEM imaging during in-situ heating.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Silicon and its composites are key materials owing to their extensive use in the semiconductor industry. While the diamond-structured form dominates, other allotropes with superior properties at ambient conditions remain of interest. Toward this, Si46 clathrate type-I crystals containing alkali/alkaline-earth metals have been extensively studied, but the experimental observation of a guest-free Si46 structure has been challenging. Using advanced electron microscopy, we show experimental evidence of guest-free clathrate-I Si46 framework from Ba8-xSi46 under in situ heating. We reveal the stepwise Ba evacuation process, starting with loss of Ba1 from the smaller cages to form Ba6Si46, followed by removal of Ba2 in larger cages to reach Si46 that appears in the thin region of the nanocrystal with a thickness around 4-6 nm at 500 °C. Calculations give a quasi-direct bandgap of 1.89 eV and support the preferential evacuation of Ba1. The observation of this guest-free Si46 framework opens up possibilities for applications in high-speed transistors, optoelectronic devices or solar cell technologies. |
| 258. | | Sheng Wu, Shunyu Wang, Zhigang Shao, Yinzhen Wang, Puxian Xiong Self-powered near-infrared mechanoluminescence through MgO/MgF2 piezo-photonic heterojunctions In: Nature Communications, vol. 16, no. 8912, 2025, (Near-infrared mechanoluminescence can be used in biostress imaging, but is subject to limitations in terms of the pre-irradiation process and low intensity. Here, the authors have circumvented these limitations by constructing an MgO/MgF2:Cr3+ heterojunction piezo-photonics system.
). @article{nokey,
title = {Self-powered near-infrared mechanoluminescence through MgO/MgF2 piezo-photonic heterojunctions},
author = {Sheng Wu and Shunyu Wang and Zhigang Shao and Yinzhen Wang and Puxian Xiong },
url = {https://www.nature.com/articles/s41467-025-63980-4.pdf},
doi = {10.1038/s41467-025-63980-4},
year = {2025},
date = {2025-10-07},
journal = {Nature Communications},
volume = {16},
number = {8912},
abstract = {Near-infrared mechanoluminescent (NIR ML) materials attract considerable attention for their force-to-light conversion capabilities. However, current materials generally have disadvantages such as high threshold and poor self-recovery ability, which limit their practical applications. Herein, we present a self-powered NIR ML material MgF2:Cr3+, which does not require pre-charging process. Leveraging the structural similarity between MgF2 and MgO, we design a MgO/MgF2:Cr3+ heterojunction piezo-photonic system that exhibits high intensity, low activation threshold, and excellent self-powered ML performance. By tuning the molar ratio of MgO to MgF2, the optimized ML intensity enhances by ≈18 times. Kelvin probe force microscopy surface potential measurement reveals a significant built-in electric field at MgF2:Cr3+ heterojunction interface. Based on the first-principle calculation results, the excellent ML performance originates from the offset of the valence band and the conduction band in the MgO/MgF2:Cr3+ heterostructure and the narrowing of the band gap, which significantly improve the electron (4.09 × 102 cm2 V-1 s-1) and hole (4.62 × 102 cm2 V-1 s-1) mobility, thereby boosting charge transfer and recombination processes. This study provides a strategy for designing high-performance self-powered NIR ML materials based on interfacial effects, offering insights into their expanded applications in the potential bio stress related biological field.},
note = {Near-infrared mechanoluminescence can be used in biostress imaging, but is subject to limitations in terms of the pre-irradiation process and low intensity. Here, the authors have circumvented these limitations by constructing an MgO/MgF2:Cr3+ heterojunction piezo-photonics system.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Near-infrared mechanoluminescent (NIR ML) materials attract considerable attention for their force-to-light conversion capabilities. However, current materials generally have disadvantages such as high threshold and poor self-recovery ability, which limit their practical applications. Herein, we present a self-powered NIR ML material MgF2:Cr3+, which does not require pre-charging process. Leveraging the structural similarity between MgF2 and MgO, we design a MgO/MgF2:Cr3+ heterojunction piezo-photonic system that exhibits high intensity, low activation threshold, and excellent self-powered ML performance. By tuning the molar ratio of MgO to MgF2, the optimized ML intensity enhances by ≈18 times. Kelvin probe force microscopy surface potential measurement reveals a significant built-in electric field at MgF2:Cr3+ heterojunction interface. Based on the first-principle calculation results, the excellent ML performance originates from the offset of the valence band and the conduction band in the MgO/MgF2:Cr3+ heterostructure and the narrowing of the band gap, which significantly improve the electron (4.09 × 102 cm2 V-1 s-1) and hole (4.62 × 102 cm2 V-1 s-1) mobility, thereby boosting charge transfer and recombination processes. This study provides a strategy for designing high-performance self-powered NIR ML materials based on interfacial effects, offering insights into their expanded applications in the potential bio stress related biological field. |
| 257. | | Zi-Feng Yuan, Ye Tian, Binze Tang, Tiancheng Liang, Chon-Hei Lo, Zixiang Yan, Dong Guan, Jiadong Guo, En-Ge Wang, Ying Jiang, Limei Xu Atomic-resolution imaging reveals nucleus-free crystallization in two-dimensional amorphous ice on graphite In: Nature Communications, vol. 16, no. 8628, 2025, (The crystallization mechanism at reduced dimensions remains poorly understood. Here, the authors use AFM and MD simulations to elucidate the nucleus-free crystallization of two-dimensional ice at atomic resolution.). @article{nokey,
title = {Atomic-resolution imaging reveals nucleus-free crystallization in two-dimensional amorphous ice on graphite},
author = {Zi-Feng Yuan and Ye Tian and Binze Tang and Tiancheng Liang and Chon-Hei Lo and Zixiang Yan and Dong Guan and Jiadong Guo and En-Ge Wang and Ying Jiang and Limei Xu },
url = {https://www.nature.com/articles/s41467-025-63664-z.pdf},
doi = {10.1038/s41467-025-63664-z},
year = {2025},
date = {2025-09-29},
journal = {Nature Communications},
volume = {16},
number = {8628},
abstract = {Two-dimensional (2D) crystallization at interfaces or in thin films plays a critical role in many natural phenomena and technological applications, yet its microscopic mechanism remains elusive due to the challenges of directly observing atomic-scale transient states during crystallization. Here, we present atomic-resolution imaging of 2D ice crystallization on graphite surface using qPlus-based cryogenic atomic force microscopy (AFM) combined with molecular dynamics (MD) simulations. The crystallization of 2D amorphous bilayer ice undergoes a fractal-to-compact transition as temperature increases. Instead of forming a critical nucleus as predicted by classical theories, the crystallization firstly proceeds via the dendritic extension of fractal islands, followed by compact growth with defect healing at the percolated edges. We find that this process is significantly assisted by out-of-plane adsorbed (ad-) water molecules, which, like a spider weaving its web, facilitate the rearrangement of hydrogen-bonding network from disordered pentagons or heptagons to ordered hexagons. This fractal-to-compact crystallization pathway, mediated by ad-molecules, presents a non-classical ordering mechanism beyond classical nucleation theory, and may offer general insights into the crystallization at the 2D limit.},
note = {The crystallization mechanism at reduced dimensions remains poorly understood. Here, the authors use AFM and MD simulations to elucidate the nucleus-free crystallization of two-dimensional ice at atomic resolution.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Two-dimensional (2D) crystallization at interfaces or in thin films plays a critical role in many natural phenomena and technological applications, yet its microscopic mechanism remains elusive due to the challenges of directly observing atomic-scale transient states during crystallization. Here, we present atomic-resolution imaging of 2D ice crystallization on graphite surface using qPlus-based cryogenic atomic force microscopy (AFM) combined with molecular dynamics (MD) simulations. The crystallization of 2D amorphous bilayer ice undergoes a fractal-to-compact transition as temperature increases. Instead of forming a critical nucleus as predicted by classical theories, the crystallization firstly proceeds via the dendritic extension of fractal islands, followed by compact growth with defect healing at the percolated edges. We find that this process is significantly assisted by out-of-plane adsorbed (ad-) water molecules, which, like a spider weaving its web, facilitate the rearrangement of hydrogen-bonding network from disordered pentagons or heptagons to ordered hexagons. This fractal-to-compact crystallization pathway, mediated by ad-molecules, presents a non-classical ordering mechanism beyond classical nucleation theory, and may offer general insights into the crystallization at the 2D limit. |
| 256. | | Jingshan S. Du, Suvo Banik, Henry Chan, Birk Fritsch, Ying Xia, Ajay S. Karakoti, Andreas Hutzler, Subramanian K. R. S. Sankaranarayanan, James J. De Yoreo Molecular-resolution imaging of ice crystallized from liquid water by cryogenic liquid-cell TEM In: Nature Communications, vol. 16, no. 8342, 2025, (Although ice plays a crucial role in shaping our ecosphere, its microstructures formed by freezing water remain unclear. Here, the authors employ cryogenic liquid-cell TEM to gain atomic-level insights into ice defects frozen from liquid water.). @article{nokey,
title = {Molecular-resolution imaging of ice crystallized from liquid water by cryogenic liquid-cell TEM},
author = {Jingshan S. Du and Suvo Banik and Henry Chan and Birk Fritsch and Ying Xia and Ajay S. Karakoti and Andreas Hutzler and Subramanian K. R. S. Sankaranarayanan and James J. De Yoreo },
url = {https://www.nature.com/articles/s41467-025-62451-0.pdf},
doi = {10.1038/s41467-025-62451-0},
year = {2025},
date = {2025-09-25},
journal = {Nature Communications},
volume = {16},
number = {8342},
abstract = {Despite the ubiquity of ice, a molecular-resolution image of nanoscopic defects or microstructures in ice crystallized from liquid water has never been obtained. This is mainly due to the difficulties in preparing and preserving crystalline ice samples that can survive under high-resolution imaging conditions. Here, we report the stabilization and Å-resolution electron imaging of ice Ih crystallized from liquid water by developing cryogenic liquid-cell transmission electron microscopy (CRYOLIC-TEM). We combine lattice mapping with molecular dynamics simulations to reveal that ice formation is highly tolerant to nanoscale defects such as misoriented subdomains and trapped gas bubbles, which are stabilized by molecular-scale structural motifs. Importantly, bubble surfaces adopt low-energy nanofacets and create negligible strain fields in the surrounding crystal. These bubbles can dynamically nucleate, grow, migrate, dissolve, and coalesce under electron irradiation and be monitored in situ near a steady state. This work improves our understanding of water crystallization behaviors at a molecular spatial resolution.},
note = {Although ice plays a crucial role in shaping our ecosphere, its microstructures formed by freezing water remain unclear. Here, the authors employ cryogenic liquid-cell TEM to gain atomic-level insights into ice defects frozen from liquid water.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Despite the ubiquity of ice, a molecular-resolution image of nanoscopic defects or microstructures in ice crystallized from liquid water has never been obtained. This is mainly due to the difficulties in preparing and preserving crystalline ice samples that can survive under high-resolution imaging conditions. Here, we report the stabilization and Å-resolution electron imaging of ice Ih crystallized from liquid water by developing cryogenic liquid-cell transmission electron microscopy (CRYOLIC-TEM). We combine lattice mapping with molecular dynamics simulations to reveal that ice formation is highly tolerant to nanoscale defects such as misoriented subdomains and trapped gas bubbles, which are stabilized by molecular-scale structural motifs. Importantly, bubble surfaces adopt low-energy nanofacets and create negligible strain fields in the surrounding crystal. These bubbles can dynamically nucleate, grow, migrate, dissolve, and coalesce under electron irradiation and be monitored in situ near a steady state. This work improves our understanding of water crystallization behaviors at a molecular spatial resolution. |
| 255. | | Lei Luo, Yao Wu, Lei Li, Zhonghan Zhang, Lu Zheng, Chao Zhu, Manzhang Xu, Weiwei Li, Ruihuan Duan, Yanchao He, Xin Zhou, Qundong Fu, Gang Wu, Jiefu Yang, Qi Wu, Wei Huang, Xuewen Wang, Zheng Liu Symmetry-broken MoS2 nanotubes through sequential sulfurization of MoO2 nanowires In: Nature Communications, vol. 16, no. 8394, 2025, (Transition metal dichalcogenide nanotubes possess symmetry-breaking properties promising for fundamental physics research. Here, the authors report a direct synthesis of crystalline MoS2 nanotubes exhibiting strong polarization and bulk photovoltaic effects.). @article{nokey,
title = {Symmetry-broken MoS2 nanotubes through sequential sulfurization of MoO2 nanowires},
author = {Lei Luo and Yao Wu and Lei Li and Zhonghan Zhang and Lu Zheng and Chao Zhu and Manzhang Xu and Weiwei Li and Ruihuan Duan and Yanchao He and Xin Zhou and Qundong Fu and Gang Wu and Jiefu Yang and Qi Wu and Wei Huang and Xuewen Wang and Zheng Liu },
url = {https://www.nature.com/articles/s41467-025-63333-1.pdf},
doi = {10.1038/s41467-025-63333-1},
year = {2025},
date = {2025-09-25},
journal = {Nature Communications},
volume = {16},
number = {8394},
abstract = {Transition metal dichalcogenide (TMD) nanotubes are emerging quantum materials with distinctive symmetry-breaking properties, offering significant potential for energy conversion technologies. However, the direct synthesis of crystalline MoS2 nanotubes remains challenging due to limited understanding of their high-temperature growth mechanisms. Here, we present a robust and controllable strategy for the direct growth of crystalline MoS2 nanotubes with well-defined tubular morphology and high structural uniformity. This approach features two key innovations: first, the controlled introduction of hydrogen reduces MoO3 into one-dimensional (1D) tetragonal MoO2 (space group I4/m) chains via a vapor–liquid–solid (VLS) mechanism; second, precise temperature zoning ensures timely sulfur vapor infusion for complete sulfurization. The intermediate MoO2 phase, with its singular crystallographic orientation, acts as an ideal template for nanotube formation. Tellurium (Te) serves as a fluxing mediator to promote the formation of uniform MoO2 nanowires, which are subsequently converted into MoS2 nanotubes. By systematically tuning the hydrogen concentration, we reveal its critical role in directing product morphology. The resulting MoS2 nanotubes exhibit pronounced symmetry breaking and significant bulk photovoltaic performance, achieving a photoresponsivity of 510 A cm−2 under 1.88 × 104 W cm−2 illumination. This work advances both the fundamental understanding of nanotube growth and the development of symmetry-engineered optoelectronic materials.},
note = {Transition metal dichalcogenide nanotubes possess symmetry-breaking properties promising for fundamental physics research. Here, the authors report a direct synthesis of crystalline MoS2 nanotubes exhibiting strong polarization and bulk photovoltaic effects.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Transition metal dichalcogenide (TMD) nanotubes are emerging quantum materials with distinctive symmetry-breaking properties, offering significant potential for energy conversion technologies. However, the direct synthesis of crystalline MoS2 nanotubes remains challenging due to limited understanding of their high-temperature growth mechanisms. Here, we present a robust and controllable strategy for the direct growth of crystalline MoS2 nanotubes with well-defined tubular morphology and high structural uniformity. This approach features two key innovations: first, the controlled introduction of hydrogen reduces MoO3 into one-dimensional (1D) tetragonal MoO2 (space group I4/m) chains via a vapor–liquid–solid (VLS) mechanism; second, precise temperature zoning ensures timely sulfur vapor infusion for complete sulfurization. The intermediate MoO2 phase, with its singular crystallographic orientation, acts as an ideal template for nanotube formation. Tellurium (Te) serves as a fluxing mediator to promote the formation of uniform MoO2 nanowires, which are subsequently converted into MoS2 nanotubes. By systematically tuning the hydrogen concentration, we reveal its critical role in directing product morphology. The resulting MoS2 nanotubes exhibit pronounced symmetry breaking and significant bulk photovoltaic performance, achieving a photoresponsivity of 510 A cm−2 under 1.88 × 104 W cm−2 illumination. This work advances both the fundamental understanding of nanotube growth and the development of symmetry-engineered optoelectronic materials. |
| 254. | | Felix Stete, Shivani Kesarwani, Charlotte Ruhmlieb, Sven H. C. Askes, Florian Schulz, Matias Bargheer, Holger Lange Inverted temperature gradients in gold–palladium antenna-reactor nanoparticles In: Nature Communications, vol. 16, no. 8168, 2025, (When light is absorbed by metal nanoparticles, electromagnetic energy is focused far beyond the diffraction limit. Here, the authors show that this energy can be further localized by the dissipation of energy within a bimetallic antenna-reactor system into attached palladium satellites.). @article{nokey,
title = {Inverted temperature gradients in gold–palladium antenna-reactor nanoparticles},
author = {Felix Stete and Shivani Kesarwani and Charlotte Ruhmlieb and Sven H. C. Askes and Florian Schulz and Matias Bargheer and Holger Lange },
url = {https://www.nature.com/articles/s41467-025-63327-z.pdf},
doi = {10.1038/s41467-025-63327-z},
year = {2025},
date = {2025-09-01},
journal = {Nature Communications},
volume = {16},
number = {8168},
abstract = {In addition to enhanced fields and possible charge transfer, the concentration of photothermal energy at the nanoscale is a central feature of plasmon-driven photochemistry. It is well known that light energy can be efficiently concentrated in metal nanoparticles to length scales far below the wavelength of light. Here we demonstrate that the energy absorbed by a gold nanoparticle can be further localized within a bimetallic gold-paladium nanoparticle system by the dissipation of energy into the attached palladium satellite nanoparticles. After pulsed excitation of the gold core, the satellites collect nearly all photothermal energy and heat up by 180 K while the light-absorbing gold core remains much colder. By comparing transient absorption dynamics of a series of bimetallic nanoparticles with a three-temperature model, we can precisely assess the temperatures of the electronic and vibrational subsystems. We find a strong inverted temperature gradient that opposes the direction of energy input and concentrates the light energy at the active catalytic nanosite.},
note = {When light is absorbed by metal nanoparticles, electromagnetic energy is focused far beyond the diffraction limit. Here, the authors show that this energy can be further localized by the dissipation of energy within a bimetallic antenna-reactor system into attached palladium satellites.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In addition to enhanced fields and possible charge transfer, the concentration of photothermal energy at the nanoscale is a central feature of plasmon-driven photochemistry. It is well known that light energy can be efficiently concentrated in metal nanoparticles to length scales far below the wavelength of light. Here we demonstrate that the energy absorbed by a gold nanoparticle can be further localized within a bimetallic gold-paladium nanoparticle system by the dissipation of energy into the attached palladium satellite nanoparticles. After pulsed excitation of the gold core, the satellites collect nearly all photothermal energy and heat up by 180 K while the light-absorbing gold core remains much colder. By comparing transient absorption dynamics of a series of bimetallic nanoparticles with a three-temperature model, we can precisely assess the temperatures of the electronic and vibrational subsystems. We find a strong inverted temperature gradient that opposes the direction of energy input and concentrates the light energy at the active catalytic nanosite. |
| 253. | | Jinyu Zhou, Xiuling Zha, Siying Ma, Sihui Wu, Chunlan Ma, Gaoyuan Chen, Zhigang Chen, Taoyang Zhang, Zhiwei Chen, Di Wang, Yuxiang Yan, Yuqing Sun, Hengdong Ren, Hongzhao Sun, Xinglong Wu, Zhigang Zhao, Shan Cong Polymer-guided grafting of single W atoms onto titanate nanotubes increases SERS activity in semiconductors In: Nature Communications, vol. 16, no. 8042, 2025, (Atomically dispersed metal atoms on semiconductor surfaces alter the local surface carrier distribution. Here, the authors demonstrate the improvement of the SERS activity of titanate nanotubes by controllable loading with monoatomic W species.
). @article{nokey,
title = {Polymer-guided grafting of single W atoms onto titanate nanotubes increases SERS activity in semiconductors},
author = {Jinyu Zhou and Xiuling Zha and Siying Ma and Sihui Wu and Chunlan Ma and Gaoyuan Chen and Zhigang Chen and Taoyang Zhang and Zhiwei Chen and Di Wang and Yuxiang Yan and Yuqing Sun and Hengdong Ren and Hongzhao Sun and Xinglong Wu and Zhigang Zhao and Shan Cong },
url = {https://www.nature.com/articles/s41467-025-63224-5.pdf},
doi = {10.1038/s41467-025-63224-5},
year = {2025},
date = {2025-08-28},
urldate = {2025-08-28},
journal = {Nature Communications},
volume = {16},
number = {8042},
abstract = {Metal single atoms have been demonstrated to induce surface-enhanced Raman scattering (SERS) due to their effectiveness in the modification of electronic structure. However, precisely modulating the relative positions of metal single atoms on sub-nanolattices remains a formidable challenge, which makes SERS studies of metal single atoms dependent on localized environments still lacking. Herein, we rely on polyethylene glycol (PEG) as a soft template to achieve the modulation of the relative positions of W atoms on titanate nanotubes (W-TNTs) and probe the local-environment-dependent SERS induced by metal single atoms based on this technique. We find that the relative position of the W single atoms greatly affects their SERS performance. This phenomenon has been attributed to the difference in charge transfer ability between single W atoms of different configurations, with isolated W atoms inducing a significantly higher density of electronic states near the Fermi energy than associated W atoms, leading to an enhanced polarization of the probe molecule and subsequently a stronger Raman signal. Our findings demonstrate a technique to effectively control the relative positions of single atoms and provide insights into single-atom-induced SERS associated with localized environments, which will facilitate the rational design of SERS substrates based on metal single atoms.},
note = {Atomically dispersed metal atoms on semiconductor surfaces alter the local surface carrier distribution. Here, the authors demonstrate the improvement of the SERS activity of titanate nanotubes by controllable loading with monoatomic W species.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Metal single atoms have been demonstrated to induce surface-enhanced Raman scattering (SERS) due to their effectiveness in the modification of electronic structure. However, precisely modulating the relative positions of metal single atoms on sub-nanolattices remains a formidable challenge, which makes SERS studies of metal single atoms dependent on localized environments still lacking. Herein, we rely on polyethylene glycol (PEG) as a soft template to achieve the modulation of the relative positions of W atoms on titanate nanotubes (W-TNTs) and probe the local-environment-dependent SERS induced by metal single atoms based on this technique. We find that the relative position of the W single atoms greatly affects their SERS performance. This phenomenon has been attributed to the difference in charge transfer ability between single W atoms of different configurations, with isolated W atoms inducing a significantly higher density of electronic states near the Fermi energy than associated W atoms, leading to an enhanced polarization of the probe molecule and subsequently a stronger Raman signal. Our findings demonstrate a technique to effectively control the relative positions of single atoms and provide insights into single-atom-induced SERS associated with localized environments, which will facilitate the rational design of SERS substrates based on metal single atoms. |
| 252. | | Yongli Ji, Caoyu Yang, Yutong Ye, Yin Zhang, Tingting Zhao, Shuyue Kong, Hongli Chen, Pai Liu, Zelong Zhao, Yilong Li, Jing Li, Ruixiao Ma, Zhiyong Ban, Kuo Yuan, Zhiyong Tang, Yi Liu, Meiting Zhao, Jun Guo Disassembly of chiral hydrogen-bonded frameworks into single-unit organometallic helices for enantioselective amyloid inhibition In: Nature Communications, vol. 16, no. 8019, 2025, (Construction of chiral nanostructures is impeded by the entropic barrier of dissymmetric blocks. Here, the authors report a dynamic assembly/disassembly strategy to yield single-unit organometallic helices for enantioselective amyloid inhibition.). @article{nokey,
title = {Disassembly of chiral hydrogen-bonded frameworks into single-unit organometallic helices for enantioselective amyloid inhibition},
author = {Yongli Ji and Caoyu Yang and Yutong Ye and Yin Zhang and Tingting Zhao and Shuyue Kong and Hongli Chen and Pai Liu and Zelong Zhao and Yilong Li and Jing Li and Ruixiao Ma and Zhiyong Ban and Kuo Yuan and Zhiyong Tang and Yi Liu and Meiting Zhao and Jun Guo },
url = {https://www.nature.com/articles/s41467-025-63459-2.pdf},
doi = {10.1038/s41467-025-63459-2},
year = {2025},
date = {2025-08-27},
urldate = {2025-08-27},
journal = {Nature Communications},
volume = {16},
number = {8019},
abstract = {Chiral nanostructures hold transformative potential across diverse fields, yet their assembly construction remains hindered by the high entropic barrier of dissymmetric building units. Inspired by biological structural dynamics, we construct two chiral copper-based hydrogen-bonded frameworks [D(L)-Cu-crystals] via hydrogen-bonded assembly using chiral metal-organic helical as the building unit. Single-crystal X-ray diffraction elucidates hierarchical chirality evolution from asymmetric coordinations to helical chains and framework packing. Furthermore, disassembling D(L)-Cu-crystals yields corresponding single-unit chiral metal-organic helices [D(L)-Cu-SMOHs], fully exposed active sites and well-preserved helical architectures. Notably, D(L)-Cu-SMOHs inhibit amyloid fibrillization effectively with pronounced chirality discrimination, driven by entropy-favored hydrophobic interaction. Molecular docking reveals that D-Cu-SMOH exhibits enhanced binding to critical amyloidogenic regions relative to the L-enantiomer. This work establishes a dynamic and reversible assembly-disassembly approach applicable for constructions of chiral nanomaterials. Moreover, it provides insights into understanding enantioselective amyloid inhibition, extending applications in asymmetric catalysis, enantioselective separation and chiroptical devices.},
note = {Construction of chiral nanostructures is impeded by the entropic barrier of dissymmetric blocks. Here, the authors report a dynamic assembly/disassembly strategy to yield single-unit organometallic helices for enantioselective amyloid inhibition.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chiral nanostructures hold transformative potential across diverse fields, yet their assembly construction remains hindered by the high entropic barrier of dissymmetric building units. Inspired by biological structural dynamics, we construct two chiral copper-based hydrogen-bonded frameworks [D(L)-Cu-crystals] via hydrogen-bonded assembly using chiral metal-organic helical as the building unit. Single-crystal X-ray diffraction elucidates hierarchical chirality evolution from asymmetric coordinations to helical chains and framework packing. Furthermore, disassembling D(L)-Cu-crystals yields corresponding single-unit chiral metal-organic helices [D(L)-Cu-SMOHs], fully exposed active sites and well-preserved helical architectures. Notably, D(L)-Cu-SMOHs inhibit amyloid fibrillization effectively with pronounced chirality discrimination, driven by entropy-favored hydrophobic interaction. Molecular docking reveals that D-Cu-SMOH exhibits enhanced binding to critical amyloidogenic regions relative to the L-enantiomer. This work establishes a dynamic and reversible assembly-disassembly approach applicable for constructions of chiral nanomaterials. Moreover, it provides insights into understanding enantioselective amyloid inhibition, extending applications in asymmetric catalysis, enantioselective separation and chiroptical devices. |
| 251. | | Pan Peng, Xinqin Liu, Shuming Yang, Renjie Zhou, Hui Deng, Liang Gao, Nicholas X. Fang, Shiyuan Liu, Jinlong Zhu Anomalous photoelectrochemical etching of undoped semiconductor surfaces In: Nature Communications, vol. 16, no. 7885, 2025, (Photoelectrochemical etching relies on light-driven carrier migration to catalyze reactions on semiconductor surfaces. Here, the authors show that lateral photon gradients induce anomalous etching of undoped semiconductor materials.). @article{nokey,
title = {Anomalous photoelectrochemical etching of undoped semiconductor surfaces},
author = {Pan Peng and Xinqin Liu and Shuming Yang and Renjie Zhou and Hui Deng and Liang Gao and Nicholas X. Fang and Shiyuan Liu and Jinlong Zhu },
url = {https://www.nature.com/articles/s41467-025-63252-1.pdf},
doi = {10.1038/s41467-025-63252-1},
year = {2025},
date = {2025-08-23},
journal = {Nature Communications},
volume = {16},
number = {7885},
abstract = {For more than 60 years, it has been widely accepted that the irradiance of the incoming light plays the most critical role in the etching effect of the photoelectrochemical etching process, which is built upon the underlying physics that photo-generated charge carriers catalyze the dissolution of n-type semiconductors. However, in this paper, we report an anomalous physical phenomenon, i.e., the spatially distributed photons with a lateral gradient could drive the lateral distribution of carriers on the surface of semiconductors, which leads to the anomalous etching phenomenon on the surface of undoped semiconductor materials during the PEC etching process. Research shows that parameters such as light intensity, light intensity gradient, and carrier diffusion length are significantly correlated with this process. This discovery provides a potential method of rapid and large-scale 3D nanomanufacturing on semiconductor materials, which holds promise for significant applications in diverse fields such as microelectronics, nanophotonics, microelectromechanical systems (MEMS), and biomedicine.},
note = {Photoelectrochemical etching relies on light-driven carrier migration to catalyze reactions on semiconductor surfaces. Here, the authors show that lateral photon gradients induce anomalous etching of undoped semiconductor materials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
For more than 60 years, it has been widely accepted that the irradiance of the incoming light plays the most critical role in the etching effect of the photoelectrochemical etching process, which is built upon the underlying physics that photo-generated charge carriers catalyze the dissolution of n-type semiconductors. However, in this paper, we report an anomalous physical phenomenon, i.e., the spatially distributed photons with a lateral gradient could drive the lateral distribution of carriers on the surface of semiconductors, which leads to the anomalous etching phenomenon on the surface of undoped semiconductor materials during the PEC etching process. Research shows that parameters such as light intensity, light intensity gradient, and carrier diffusion length are significantly correlated with this process. This discovery provides a potential method of rapid and large-scale 3D nanomanufacturing on semiconductor materials, which holds promise for significant applications in diverse fields such as microelectronics, nanophotonics, microelectromechanical systems (MEMS), and biomedicine. |
| 250. | | Jalmar Tschakert, Qigang Zhong, Alexander Sekels, Pascal Henkel, Jannis Jung, K. Linus H. Pohl, Hermann A. Wegner, Doreen Mollenhauer, André Schirmeisen, Daniel Ebeling Probing weak chemical interactions of metal surface atoms with CO-terminated AFM tips identifies molecular adsorption sites In: Nature Communications, vol. 16, no. 7874, 2025, (Understanding molecule-surface interactions is key to neat on-surface synthesis of functional nanomaterials. Here, the authors measure interactions of metal surface atoms with CO-terminated tips to reveal their nature and reliable adsorption sites.). @article{nokey,
title = {Probing weak chemical interactions of metal surface atoms with CO-terminated AFM tips identifies molecular adsorption sites},
author = {Jalmar Tschakert and Qigang Zhong and Alexander Sekels and Pascal Henkel and Jannis Jung and K. Linus H. Pohl and Hermann A. Wegner and Doreen Mollenhauer and André Schirmeisen and Daniel Ebeling },
url = {https://www.nature.com/articles/s41467-025-63159-x.pdf},
doi = {10.1038/s41467-025-63159-x},
year = {2025},
date = {2025-08-23},
journal = {Nature Communications},
volume = {16},
number = {7874},
abstract = {Metal surfaces play a key role in on-surface synthesis as they provide a two-dimensional catalytic reaction environment that stimulates activation, diffusion, and coupling of molecular reactants. Fundamental understanding of the interactions between surface atoms and reactants is very limited but would enable controlling on-surface reaction processes for designing functional nanomaterials. Here, we measure chemical interactions between CO-terminated tips and Cu(111), Ag(111), and Au(111) surface atoms in all spatial directions with picometer resolution via low temperature atomic force microscopy. This allows a site-specific quantification of the weak chemical interactions of densely packed metal surface atoms and provides a picture of the potential energy landscape experienced by adsorbed reactants. Accompanying density functional theory calculations and the crystal orbital overlap population method reveal small covalent binding contributions from orbital overlap of the corresponding p- and d-states of the CO tip and the metal surface atoms as the cause for the site-specific interactions. Accessing such small covalent bonding contributions in the background of the dispersion-dominated interaction enables revealing insights into the nature of chemical bond formation with metal surface atoms and a reliable determination of molecular adsorption sites. The latter can serve both as a starting point and as a direct comparison with theoretical studies.},
note = {Understanding molecule-surface interactions is key to neat on-surface synthesis of functional nanomaterials. Here, the authors measure interactions of metal surface atoms with CO-terminated tips to reveal their nature and reliable adsorption sites.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Metal surfaces play a key role in on-surface synthesis as they provide a two-dimensional catalytic reaction environment that stimulates activation, diffusion, and coupling of molecular reactants. Fundamental understanding of the interactions between surface atoms and reactants is very limited but would enable controlling on-surface reaction processes for designing functional nanomaterials. Here, we measure chemical interactions between CO-terminated tips and Cu(111), Ag(111), and Au(111) surface atoms in all spatial directions with picometer resolution via low temperature atomic force microscopy. This allows a site-specific quantification of the weak chemical interactions of densely packed metal surface atoms and provides a picture of the potential energy landscape experienced by adsorbed reactants. Accompanying density functional theory calculations and the crystal orbital overlap population method reveal small covalent binding contributions from orbital overlap of the corresponding p- and d-states of the CO tip and the metal surface atoms as the cause for the site-specific interactions. Accessing such small covalent bonding contributions in the background of the dispersion-dominated interaction enables revealing insights into the nature of chemical bond formation with metal surface atoms and a reliable determination of molecular adsorption sites. The latter can serve both as a starting point and as a direct comparison with theoretical studies. |
| 249. | | Yaxuan Zhang, Kai Qu, Ting Pan, Yaqi Zhang, Leng Wang, Hongliang Chen Highly conductive single-molecule junctions through electrocatalytic formation of benzyl-type Au‒C bonds In: Nature Communications, vol. 16, no. 7692, 2025, (Creating robust molecular devices requires covalent electrode-molecule junctions. Here, the authors develop an electrocatalytic method using Katritzky salts to form stable Au–C bonds that exhibit high conductivity and improved robustness.). @article{nokey,
title = {Highly conductive single-molecule junctions through electrocatalytic formation of benzyl-type Au‒C bonds},
author = {Yaxuan Zhang and Kai Qu and Ting Pan and Yaqi Zhang and Leng Wang and Hongliang Chen },
url = {https://www.nature.com/articles/s41467-025-62961-x.pdf},
doi = {10.1038/s41467-025-62961-x},
year = {2025},
date = {2025-08-18},
journal = {Nature Communications},
volume = {16},
number = {7692},
abstract = {Creating reliable molecular-scale electronic devices demands strong, stable connections between metal electrodes and organic molecules. A significant challenge is forming robust chemical bonds directly to gold electrodes, as gold is notoriously unreactive. Conventional methods for creating gold-carbon (Au‒C) bonds are therefore limited. Here we demonstrate an electrocatalytic solution: using an applied voltage, we inject a single electron from a gold electrode into specific organic salts (pyridinium ions). This electron transfer breaks the salt apart, generating highly reactive carbon-based radicals. These radicals spontaneously form strong, direct covalent bonds (Au‒C) with the gold surface. Using precise single-molecule measurements, we show this radical-mediated bonding creates exceptionally stable molecular junctions. Furthermore, these junctions exhibit excellent electrical conductivity across the molecule’s core structure. This high conductivity arises because the direct Au‒C bond allows efficient overlap of electron orbitals between the gold and the molecule. Our strategy provides a versatile and controlled way to build atomically precise, highly conductive interfaces between metals and organic components, advancing the design of functional molecular electronics through tailored covalent connections.},
note = {Creating robust molecular devices requires covalent electrode-molecule junctions. Here, the authors develop an electrocatalytic method using Katritzky salts to form stable Au–C bonds that exhibit high conductivity and improved robustness.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Creating reliable molecular-scale electronic devices demands strong, stable connections between metal electrodes and organic molecules. A significant challenge is forming robust chemical bonds directly to gold electrodes, as gold is notoriously unreactive. Conventional methods for creating gold-carbon (Au‒C) bonds are therefore limited. Here we demonstrate an electrocatalytic solution: using an applied voltage, we inject a single electron from a gold electrode into specific organic salts (pyridinium ions). This electron transfer breaks the salt apart, generating highly reactive carbon-based radicals. These radicals spontaneously form strong, direct covalent bonds (Au‒C) with the gold surface. Using precise single-molecule measurements, we show this radical-mediated bonding creates exceptionally stable molecular junctions. Furthermore, these junctions exhibit excellent electrical conductivity across the molecule’s core structure. This high conductivity arises because the direct Au‒C bond allows efficient overlap of electron orbitals between the gold and the molecule. Our strategy provides a versatile and controlled way to build atomically precise, highly conductive interfaces between metals and organic components, advancing the design of functional molecular electronics through tailored covalent connections. |
| 248. | | Ya-Jie Wang, Ke-Xin Zheng, Yi-He Yu, Zhengkun Xie, Pengyao Xing, Shuang-Quan Zang Self-assembled metal clustersomes and chirality transfer to colloidal photonic crystals In: Nature Communications, vol. 16, no. 7450, 2025, (Engineering soft materials from rigid metal clusters is a challenge. Here, the authors create self-assembled metal clustersomes that combine rigid and soft characteristics for tunable photonic crystals and chiral optics.). @article{nokey,
title = {Self-assembled metal clustersomes and chirality transfer to colloidal photonic crystals},
author = {Ya-Jie Wang and Ke-Xin Zheng and Yi-He Yu and Zhengkun Xie and Pengyao Xing and Shuang-Quan Zang },
url = {https://www.nature.com/articles/s41467-025-62816-5.pdf},
doi = {10.1038/s41467-025-62816-5},
year = {2025},
date = {2025-08-12},
journal = {Nature Communications},
volume = {16},
number = {7450},
abstract = {The construction of soft and flexible materials using metallic hard synthons is expected to endow these materials with convergent properties for multiple functionalities. However, the design protocols remain underdeveloped due to limited understanding of structure-property correlations and growth preferences regarding dimensions and size. Herein, through bottom-up self-assembly of engineered atomically precise metal clusters, membranous liposomal nanoarchitectures with soft matter morphology and rigid properties are constructed. Post-modification of surface ligands introduces considerable steric effects, thereby directing self-assembly towards membranous curvature in liposomal architectures rather than crystallization. Host‒guest complexation by β-cyclodextrin enables precise regulation of their size and dispersity. Chirality can be facilely implanted to afford chiroptical properties at both ground and photoexcited states. The inherent shape-persistent and rigid properties from metal clusters confer prominent mechanical strength, with Young’s moduli reaching up to 16‒20 GPa. The characteristic of combining rigidity with soft matter morphology offers opportunities to explore application potentials in colloidal photonic crystals, particularly those with modulated structural colors. Compared with traditional liposomes and polymersomes, metal clustersomes could provide expanded functionalities.},
note = {Engineering soft materials from rigid metal clusters is a challenge. Here, the authors create self-assembled metal clustersomes that combine rigid and soft characteristics for tunable photonic crystals and chiral optics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The construction of soft and flexible materials using metallic hard synthons is expected to endow these materials with convergent properties for multiple functionalities. However, the design protocols remain underdeveloped due to limited understanding of structure-property correlations and growth preferences regarding dimensions and size. Herein, through bottom-up self-assembly of engineered atomically precise metal clusters, membranous liposomal nanoarchitectures with soft matter morphology and rigid properties are constructed. Post-modification of surface ligands introduces considerable steric effects, thereby directing self-assembly towards membranous curvature in liposomal architectures rather than crystallization. Host‒guest complexation by β-cyclodextrin enables precise regulation of their size and dispersity. Chirality can be facilely implanted to afford chiroptical properties at both ground and photoexcited states. The inherent shape-persistent and rigid properties from metal clusters confer prominent mechanical strength, with Young’s moduli reaching up to 16‒20 GPa. The characteristic of combining rigidity with soft matter morphology offers opportunities to explore application potentials in colloidal photonic crystals, particularly those with modulated structural colors. Compared with traditional liposomes and polymersomes, metal clustersomes could provide expanded functionalities. |
| 247. | | Zibing Wang, Zifeng Yuan, Mouyang Cheng, Xudan Huang, Keyang Liu, Yihan Wang, Huacong Sun, Lei Liao, Zhi Xu, Ji Chen, Wenlong Wang, Lei Liu, Xuedong Bai, Limei Xu, Enge Wang, Lifen Wang Molecularly resolved mapping of heterogeneous ice nucleation and crystallization pathways using in-situ cryo-TEM In: Nature Communications, vol. 16, no. 7349, 2025, (Understanding ice crystallization at the molecular level remains a challenge. Here, the authors use graphene-based in-situ cryo-TEM to show how vapor deposition leads to polymorphic ice formation controlled by surface energy.). @article{nokey,
title = {Molecularly resolved mapping of heterogeneous ice nucleation and crystallization pathways using in-situ cryo-TEM},
author = {Zibing Wang and Zifeng Yuan and Mouyang Cheng and Xudan Huang and Keyang Liu and Yihan Wang and Huacong Sun and Lei Liao and Zhi Xu and Ji Chen and Wenlong Wang and Lei Liu and Xuedong Bai and Limei Xu and Enge Wang and Lifen Wang },
url = {https://www.nature.com/articles/s41467-025-62900-w.pdf},
doi = {10.1038/s41467-025-62900-w},
year = {2025},
date = {2025-08-09},
journal = {Nature Communications},
volume = {16},
number = {7349},
abstract = {Crystallization plays a fundamental role in diverse fields such as glaciology, geology, biology, and materials science. Among various crystallizing systems, the formation of ice remains elusive, despite decades of intensive investigation. In this study, we integrate in-situ cryogenic transmission electron microscopy with molecular dynamics simulations to develop a molecular-resolution mapping and thermodynamic framework for deposition freezing under low-temperature, low-pressure conditions. Our results demonstrate that ice formation on rapidly cooled substrates, representing far-from-equilibrium states, proceeds via an adsorption-mediated, barrierless pathway of heterogeneous ice nucleation, followed by progression toward thermodynamic equilibrium. This process is unveiled to involve a series of distinct yet interconnected steps, including amorphous ice adsorption, spontaneous nucleation and growth of ice I, Ostwald ripening, Wulff construction, oriented coalescence, and aggregation, all governed by interfacial free energy minima. Our findings offer direct molecular-level insight into the mechanisms of heterogeneous ice nucleation, enrich current understanding of non-classical nucleation pathways, and emphasize the critical role of interfacial energetics in shaping ice crystal morphology and quality.},
note = {Understanding ice crystallization at the molecular level remains a challenge. Here, the authors use graphene-based in-situ cryo-TEM to show how vapor deposition leads to polymorphic ice formation controlled by surface energy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Crystallization plays a fundamental role in diverse fields such as glaciology, geology, biology, and materials science. Among various crystallizing systems, the formation of ice remains elusive, despite decades of intensive investigation. In this study, we integrate in-situ cryogenic transmission electron microscopy with molecular dynamics simulations to develop a molecular-resolution mapping and thermodynamic framework for deposition freezing under low-temperature, low-pressure conditions. Our results demonstrate that ice formation on rapidly cooled substrates, representing far-from-equilibrium states, proceeds via an adsorption-mediated, barrierless pathway of heterogeneous ice nucleation, followed by progression toward thermodynamic equilibrium. This process is unveiled to involve a series of distinct yet interconnected steps, including amorphous ice adsorption, spontaneous nucleation and growth of ice I, Ostwald ripening, Wulff construction, oriented coalescence, and aggregation, all governed by interfacial free energy minima. Our findings offer direct molecular-level insight into the mechanisms of heterogeneous ice nucleation, enrich current understanding of non-classical nucleation pathways, and emphasize the critical role of interfacial energetics in shaping ice crystal morphology and quality. |
| 246. | | Henrik Klein Moberg, Giuseppe Abbondanza, Ievgen Nedrygailov, David Albinsson, Joachim Fritzsche, Christoph Langhammer Deep-learning-enabled online mass spectrometry of the reaction product of a single catalyst nanoparticle In: Nature Communications, vol. 16, no. 7203, 2025, (Extracting signals from noise is challenging in single-particle catalysis. Here, the authors use deep learning to enhance mass spectrometry, enabling detection of reactions on single nanoparticles.). @article{nokey,
title = {Deep-learning-enabled online mass spectrometry of the reaction product of a single catalyst nanoparticle},
author = {Henrik Klein Moberg and Giuseppe Abbondanza and Ievgen Nedrygailov and David Albinsson and Joachim Fritzsche and Christoph Langhammer },
url = {https://www.nature.com/articles/s41467-025-62602-3.pdf},
doi = {10.1038/s41467-025-62602-3},
year = {2025},
date = {2025-08-05},
journal = {Nature Communications},
volume = {16},
number = {7203},
abstract = {Extracting weak signals from noise is a generic challenge in experimental science. In catalysis, it manifests itself as the need to quantify chemical reactions on nanoscopic surface areas, such as single nanoparticles or even single atoms. Here, we address this challenge by combining the ability of nanofluidic reactors to focus reaction product from tiny catalyst surfaces towards online mass spectrometric analysis with the high capacity of a constrained denoising auto-encoder to discern weak signals from noise. Using CO oxidation and C2H4 hydrogenation on Pd as model reactions, we demonstrate that the catalyst surface area required for online mass spectrometry can be reduced by ≈ 3 orders of magnitude compared to state of the art, down to a single nanoparticle with 0.0072 ± 0.00086 μm2 surface area. These results advocate deep learning to improve resolution in mass spectrometry in general and for online reaction analysis in single-particle catalysis in particular.},
note = {Extracting signals from noise is challenging in single-particle catalysis. Here, the authors use deep learning to enhance mass spectrometry, enabling detection of reactions on single nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Extracting weak signals from noise is a generic challenge in experimental science. In catalysis, it manifests itself as the need to quantify chemical reactions on nanoscopic surface areas, such as single nanoparticles or even single atoms. Here, we address this challenge by combining the ability of nanofluidic reactors to focus reaction product from tiny catalyst surfaces towards online mass spectrometric analysis with the high capacity of a constrained denoising auto-encoder to discern weak signals from noise. Using CO oxidation and C2H4 hydrogenation on Pd as model reactions, we demonstrate that the catalyst surface area required for online mass spectrometry can be reduced by ≈ 3 orders of magnitude compared to state of the art, down to a single nanoparticle with 0.0072 ± 0.00086 μm2 surface area. These results advocate deep learning to improve resolution in mass spectrometry in general and for online reaction analysis in single-particle catalysis in particular. |
| 245. | | Hyojung Kang, Yoojung Jeon, Kyungnae Baek, SeonJu Park, Jayoon Lee, Tae Soup Shim, Jerome K. Hyun, So-Jung Park Dynamic birefringence and chirality of magnetically controllable assemblies of anisotropic plasmonic nanoparticles in dispersion In: Nature Communications, vol. 16, no. 7076, 2025, (The ability to control the arrangement of plasmonic nanoparticles offers a promising approach to modulating optical properties. Here, the authors report on magnetically controllable assemblies of plasmonic nanoparticles in dispersion that exhibit dynamic birefringence and chirality.). @article{nokey,
title = {Dynamic birefringence and chirality of magnetically controllable assemblies of anisotropic plasmonic nanoparticles in dispersion},
author = {Hyojung Kang and Yoojung Jeon and Kyungnae Baek and SeonJu Park and Jayoon Lee and Tae Soup Shim and Jerome K. Hyun and So-Jung Park },
url = {https://www.nature.com/articles/s41467-025-62508-0.pdf},
doi = {10.1038/s41467-025-62508-0},
year = {2025},
date = {2025-08-01},
journal = {Nature Communications},
volume = {16},
number = {7076},
abstract = {The ability to control the orientation and arrangement of plasmonic nanoparticles with shape anisotropy offers a promising route to achieving highly tunable optical properties. In this study, we introduce a synthetic approach for magnetically controllable plasmonic nanoparticles (MPs) consisting of an anisotropic gold core encapsulated by an iron oxide shell. The superparamagnetic property of the iron oxide shell enables rapid, reversible, and remotely controlled alignment of MPs, allowing for dynamic manipulation of their optical properties. Linearly aligned MPs demonstrate tunable transmission colors via plasmon-mediated birefringence. Helical MP arrays exhibit circular dichroism of up to 12° and g-factors reaching 0.21—the highest reported value for solution-phase assemblies of achiral nanoparticles. The synthetic method is applicable to nanoparticles of various sizes and shapes, highlighting its generality and expandability.},
note = {The ability to control the arrangement of plasmonic nanoparticles offers a promising approach to modulating optical properties. Here, the authors report on magnetically controllable assemblies of plasmonic nanoparticles in dispersion that exhibit dynamic birefringence and chirality.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The ability to control the orientation and arrangement of plasmonic nanoparticles with shape anisotropy offers a promising route to achieving highly tunable optical properties. In this study, we introduce a synthetic approach for magnetically controllable plasmonic nanoparticles (MPs) consisting of an anisotropic gold core encapsulated by an iron oxide shell. The superparamagnetic property of the iron oxide shell enables rapid, reversible, and remotely controlled alignment of MPs, allowing for dynamic manipulation of their optical properties. Linearly aligned MPs demonstrate tunable transmission colors via plasmon-mediated birefringence. Helical MP arrays exhibit circular dichroism of up to 12° and g-factors reaching 0.21—the highest reported value for solution-phase assemblies of achiral nanoparticles. The synthetic method is applicable to nanoparticles of various sizes and shapes, highlighting its generality and expandability. |
| 244. | | Kaustav Das, Ishita Ghosh, Soumalya Chakraborty, Amritha G. Nambiar, Sourav Maji, Sumanta K. Karan, Dinesh Kumar, Arvind K. Bansal, C. Malla Reddy Structural origin of fracture-induced surface charges in piezoelectric pharmaceutical crystals for engineering bulk properties In: Nature Communications, vol. 16, no. 6858, 2025, (Surface charging in crystalline materials is a complex phenomenon to predict and design. Here the authors show the origin of surface charges in polar piezoelectric pharmaceuticals to predict the outcome of mechanical impact on their bulk properties.). @article{nokey,
title = {Structural origin of fracture-induced surface charges in piezoelectric pharmaceutical crystals for engineering bulk properties},
author = {Kaustav Das and Ishita Ghosh and Soumalya Chakraborty and Amritha G. Nambiar and Sourav Maji and Sumanta K. Karan and Dinesh Kumar and Arvind K. Bansal and C. Malla Reddy },
url = {https://www.nature.com/articles/s41467-025-58138-1.pdf},
doi = {10.1038/s41467-025-58138-1},
year = {2025},
date = {2025-07-25},
urldate = {2025-07-25},
journal = {Nature Communications},
volume = {16},
number = {6858},
abstract = {Altering surface chemistry of functional materials is an attractive route to enable large property enhancements without sacrificing overall structural-order, appealing to diverse fields of application sciences; however, the same remains unexplored for organic crystalline materials. Herein, piezoelectricity in pharmaceutical crystals is reported to show colossal surface charges driven by mechanical fracture — where a collection of dipoles arranged in polar head-to-tail fashion generates opposite surface charges on freshly fractured faces — causing them to actuate large distances over 75 µm in milliseconds. Kelvin probe force microscopy is leveraged to show many-fold surface potential enhancement in fractured surfaces relative to the pristine crystals. Further, complementarity of the surface potentials in a pair of fractured crystal shards and asymptotic decay behaviour with time are observed. Newly formed surfaces of the pharmaceutical crystals show long-lasting charges despite their relatively lower piezo-response confirmed by bulk piezometry. To establish the generality of surface phenomena, statistical analyses (≈50 samples) of post-fracture-attraction behaviour of crystals are performed. Finally, the application of fracture-driven surface charges in industrial processes is achieved by investigating flow-property and tablet-strength of bulk pharmaceutical materials. This multiscale approach unveils the symmetry-dependency of surface charges in fractured materials, and probes the same for utilisation in bulk-property engineering.},
note = {Surface charging in crystalline materials is a complex phenomenon to predict and design. Here the authors show the origin of surface charges in polar piezoelectric pharmaceuticals to predict the outcome of mechanical impact on their bulk properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Altering surface chemistry of functional materials is an attractive route to enable large property enhancements without sacrificing overall structural-order, appealing to diverse fields of application sciences; however, the same remains unexplored for organic crystalline materials. Herein, piezoelectricity in pharmaceutical crystals is reported to show colossal surface charges driven by mechanical fracture — where a collection of dipoles arranged in polar head-to-tail fashion generates opposite surface charges on freshly fractured faces — causing them to actuate large distances over 75 µm in milliseconds. Kelvin probe force microscopy is leveraged to show many-fold surface potential enhancement in fractured surfaces relative to the pristine crystals. Further, complementarity of the surface potentials in a pair of fractured crystal shards and asymptotic decay behaviour with time are observed. Newly formed surfaces of the pharmaceutical crystals show long-lasting charges despite their relatively lower piezo-response confirmed by bulk piezometry. To establish the generality of surface phenomena, statistical analyses (≈50 samples) of post-fracture-attraction behaviour of crystals are performed. Finally, the application of fracture-driven surface charges in industrial processes is achieved by investigating flow-property and tablet-strength of bulk pharmaceutical materials. This multiscale approach unveils the symmetry-dependency of surface charges in fractured materials, and probes the same for utilisation in bulk-property engineering. |
| 243. | | Qingbin Zeng, Quer Yue, Ming Zhang, Rui Zhou, Zhen Wang, Zhongyu Xiao, Yaping Yuan, Xiuchao Zhao, Weiping Jiang, Lei Zhang, Yuqi Yang, Minghui Yang, Haidong Li, Louis-S. Bouchard, Qianni Guo, Xin Zhou Multivariate metal-organic frameworks enable chemical shift-encoded MRI with femtomolar sensitivity for biological systems In: Nature Communications, vol. 16, no. 6832, 2025, (Conventional entrapped 129Xe with low signal intensity limits in vivo applications. Here, the authors developed a MOF-based reporter that enhances signal intensity, achieves femtomolar detection, and enables chemical shift-encoded MRI in rat lungs.). @article{nokey,
title = {Multivariate metal-organic frameworks enable chemical shift-encoded MRI with femtomolar sensitivity for biological systems},
author = {Qingbin Zeng and Quer Yue and Ming Zhang and Rui Zhou and Zhen Wang and Zhongyu Xiao and Yaping Yuan and Xiuchao Zhao and Weiping Jiang and Lei Zhang and Yuqi Yang and Minghui Yang and Haidong Li and Louis-S. Bouchard and Qianni Guo and Xin Zhou },
url = {https://www.nature.com/articles/s41467-025-62110-4.pdf},
doi = {10.1038/s41467-025-62110-4},
year = {2025},
date = {2025-07-24},
journal = {Nature Communications},
volume = {16},
number = {6832},
abstract = {Fluorescent molecules with specific target moieties are essential for histopathological analysis, but their limited tissue penetration depth makes in vivo, in situ color encoding analysis challenging. Magnetic resonance imaging (MRI) offers deep tissue penetration. When combined with chemical shift-encoded MRI reporters, it enables in vivo chemical shift encoding for biotarget imaging and analysis. These reporters require both strong signal intensity and large chemical shift window. However, conventional proton MRI reporters, with low sensitivity and a small chemical shift window, limit their in vivo applications. Here, we describe a chemical shift-encoded hyperpolarized 129Xe MRI reporter based on the multivariate metal-organic framework, NiZn-ZIF-8, to overcome these challenges. The proposed NiZn-ZIF-8 gives distinct chemical shifts for dissolved and entrapped 129Xe without signal interference, enhancing the 129Xe NMR signal by 210 times compared to dissolved 129Xe in water and biological media. This enables detection threshold at ≈ 4 fM concentrations, setting a record for the lowest concentration of xenon hosts detected in nanomaterials. Additionally, NiZn-ZIF-8 exhibits good in vivo MRI performance, allowing xenon encoding and distinction in rat lungs. NiZn-ZIF-8 represents a versatile and powerful platform for advanced molecular imaging and in vivo biomedical diagnostics.},
note = {Conventional entrapped 129Xe with low signal intensity limits in vivo applications. Here, the authors developed a MOF-based reporter that enhances signal intensity, achieves femtomolar detection, and enables chemical shift-encoded MRI in rat lungs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fluorescent molecules with specific target moieties are essential for histopathological analysis, but their limited tissue penetration depth makes in vivo, in situ color encoding analysis challenging. Magnetic resonance imaging (MRI) offers deep tissue penetration. When combined with chemical shift-encoded MRI reporters, it enables in vivo chemical shift encoding for biotarget imaging and analysis. These reporters require both strong signal intensity and large chemical shift window. However, conventional proton MRI reporters, with low sensitivity and a small chemical shift window, limit their in vivo applications. Here, we describe a chemical shift-encoded hyperpolarized 129Xe MRI reporter based on the multivariate metal-organic framework, NiZn-ZIF-8, to overcome these challenges. The proposed NiZn-ZIF-8 gives distinct chemical shifts for dissolved and entrapped 129Xe without signal interference, enhancing the 129Xe NMR signal by 210 times compared to dissolved 129Xe in water and biological media. This enables detection threshold at ≈ 4 fM concentrations, setting a record for the lowest concentration of xenon hosts detected in nanomaterials. Additionally, NiZn-ZIF-8 exhibits good in vivo MRI performance, allowing xenon encoding and distinction in rat lungs. NiZn-ZIF-8 represents a versatile and powerful platform for advanced molecular imaging and in vivo biomedical diagnostics. |
| 242. | | Dylan Jennings, Moritz L. Weber, Ansgar Meise, Tobias Binninger, Conor J. Price, Moritz Kindelmann, Ivar Reimanis, Hiroaki Matsumoto, Pengfei Cao, Regina Dittmann, Piotr M. Kowalski, Marc Heggen, Olivier Guillon, Joachim Mayer, Felix Gunkel, Wolfgang Rheinheimer Direct atomic-scale investigation of the coarsening mechanisms of exsolved catalytic Ni nanoparticles In: Nature Communications, vol. 16, no. 6830, 2025, (Exsolution enables the formation of active catalytic nanoparticles, but their thermal stability remains limited. Here, the authors use secondary-electron imaging at atomic resolution to directly visualize the coarsening of exsolved nanoparticles.). @article{nokey,
title = {Direct atomic-scale investigation of the coarsening mechanisms of exsolved catalytic Ni nanoparticles},
author = {Dylan Jennings and Moritz L. Weber and Ansgar Meise and Tobias Binninger and Conor J. Price and Moritz Kindelmann and Ivar Reimanis and Hiroaki Matsumoto and Pengfei Cao and Regina Dittmann and Piotr M. Kowalski and Marc Heggen and Olivier Guillon and Joachim Mayer and Felix Gunkel and Wolfgang Rheinheimer },
url = {https://www.nature.com/articles/s41467-025-61971-z.pdf},
doi = {10.1038/s41467-025-61971-z},
year = {2025},
date = {2025-07-24},
journal = {Nature Communications},
volume = {16},
number = {6830},
abstract = {Exsolution-active catalysts allow for the formation of highly active metallic nanoparticles, yet recent work has shown that their long-term thermal stability remains a challenge. In this work, the dynamics of exsolved Ni nanoparticles are probed in-situ with atomically resolved secondary electron imaging with environmental scanning transmission electron microscopy. Pre-characterization shows embedded NiOx nanostructures within the parent oxide. Subsequent in-situ exsolution demonstrates that two populations of exsolved particles form with distinct metal-support interactions and coarsening behaviors. Nanoparticles which precipitate above embedded nanostructures are observed to be more stable, and are prevented from migrating on the surface of the support. Nanoparticle migration which fits random-walk kinetics is observed, and particle behavior is shown to be analogous to a classical wetting model. Additionally, DFT calculations indicate that particle motion is facilitated by the support oxide. Ostwald ripening processes are visualized simultaneously to migration, including particle redissolution and particle ripening.},
note = {Exsolution enables the formation of active catalytic nanoparticles, but their thermal stability remains limited. Here, the authors use secondary-electron imaging at atomic resolution to directly visualize the coarsening of exsolved nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Exsolution-active catalysts allow for the formation of highly active metallic nanoparticles, yet recent work has shown that their long-term thermal stability remains a challenge. In this work, the dynamics of exsolved Ni nanoparticles are probed in-situ with atomically resolved secondary electron imaging with environmental scanning transmission electron microscopy. Pre-characterization shows embedded NiOx nanostructures within the parent oxide. Subsequent in-situ exsolution demonstrates that two populations of exsolved particles form with distinct metal-support interactions and coarsening behaviors. Nanoparticles which precipitate above embedded nanostructures are observed to be more stable, and are prevented from migrating on the surface of the support. Nanoparticle migration which fits random-walk kinetics is observed, and particle behavior is shown to be analogous to a classical wetting model. Additionally, DFT calculations indicate that particle motion is facilitated by the support oxide. Ostwald ripening processes are visualized simultaneously to migration, including particle redissolution and particle ripening. |
| 241. | | Wen Zhang, Wei Zheng, Ping Huang, Dengfeng Yang, Zhiqing Shao, Wei Zhang, Hao Zhang, Zhi Xie, Jin Xu, Xueyuan Chen Intense upconverted ultraviolet emission of Er3+ through confined energy transfer in Yb3+/Er3+ co-doped Rb3InCl6 In: Nature Communications, vol. 16, no. 6762, 2025, (Intense upconverted ultraviolet emission of Er3+ is achieved through confined energy transfer in Yb3+/Er3+ co-doped 0D Rb3InCl6, enabling NIR-driven anion exchange of CsPbX3 perovskite nanocrystals in haloalkanes with high controllability.). @article{nokey,
title = {Intense upconverted ultraviolet emission of Er3+ through confined energy transfer in Yb3+/Er3+ co-doped Rb3InCl6},
author = {Wen Zhang and Wei Zheng and Ping Huang and Dengfeng Yang and Zhiqing Shao and Wei Zhang and Hao Zhang and Zhi Xie and Jin Xu and Xueyuan Chen },
url = {https://www.nature.com/articles/s41467-025-58901-4.pdf},
doi = {10.1038/s41467-025-58901-4},
year = {2025},
date = {2025-07-22},
journal = {Nature Communications},
volume = {16},
number = {6762},
abstract = {Yb3+/Er3+ activated upconversion (UC) materials have been widely applied in many advanced technologies owing to their high UC efficiency in the visible region. However, it is challenging to achieve efficient ultraviolet (UV) UC luminescence (UCL) in Yb3+/Er3+ system, due to the dense energy levels of Er3+ that impose deleterious nonradiative relaxation. Herein, we report a strategy to liberate the UV-UCL of Er3+ based on the confined energy transfer in Yb3+/Er3+ co-doped 0D Rb3InCl6 with a low phonon energy and a large interionic distance. This facilitates the population of Er3+ at the 4G11/2 state, which yields intense upconverted UV emission at 384 nm, with a much higher UV-to-green ratio (I384/I554 = 0.864) than that of traditional UC materials ( < 0.1). By leveraging the intense upconverted UV emission of Er3+, we demonstrate the application of Rb3InCl6:Yb3+/Er3+ nanocrystals as a NIR-to-UV transducer for NIR-triggered anion exchange of CsPbX3 perovskite nanocrystals with high efficiency and good controllability. These findings offer an approach for the exploration of novel UC materials via energy transfer and crystal lattice engineering towards versatile applications.},
note = {Intense upconverted ultraviolet emission of Er3+ is achieved through confined energy transfer in Yb3+/Er3+ co-doped 0D Rb3InCl6, enabling NIR-driven anion exchange of CsPbX3 perovskite nanocrystals in haloalkanes with high controllability.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Yb3+/Er3+ activated upconversion (UC) materials have been widely applied in many advanced technologies owing to their high UC efficiency in the visible region. However, it is challenging to achieve efficient ultraviolet (UV) UC luminescence (UCL) in Yb3+/Er3+ system, due to the dense energy levels of Er3+ that impose deleterious nonradiative relaxation. Herein, we report a strategy to liberate the UV-UCL of Er3+ based on the confined energy transfer in Yb3+/Er3+ co-doped 0D Rb3InCl6 with a low phonon energy and a large interionic distance. This facilitates the population of Er3+ at the 4G11/2 state, which yields intense upconverted UV emission at 384 nm, with a much higher UV-to-green ratio (I384/I554 = 0.864) than that of traditional UC materials ( < 0.1). By leveraging the intense upconverted UV emission of Er3+, we demonstrate the application of Rb3InCl6:Yb3+/Er3+ nanocrystals as a NIR-to-UV transducer for NIR-triggered anion exchange of CsPbX3 perovskite nanocrystals with high efficiency and good controllability. These findings offer an approach for the exploration of novel UC materials via energy transfer and crystal lattice engineering towards versatile applications. |
| 240. | | Changxing Wang, Yayun Ning, Yifan Yue, Guoli Du, Yuechi Xie, Jianing Li, Nazia Bibi, Xiaoxiang Wen, Jianing Li, Sen Yang, Xuegang Lu
Scalable synthesis of phosphorescent SiO2 nanospheres and their use for angle-dependent and thermoresponsive photonic gels with multimode luminescence In: Nature Communications, vol. 16, no. 6640, 2025, (Room temperature phosphorescent (RTP) materials are promising for optical applications. Here, the authors demonstrate a scalable preparation of RTP nanospheres and their assembly into photonic crystals with multimodal luminescence.). @article{nokey,
title = {Scalable synthesis of phosphorescent SiO2 nanospheres and their use for angle-dependent and thermoresponsive photonic gels with multimode luminescence},
author = {Changxing Wang and Yayun Ning and Yifan Yue and Guoli Du and Yuechi Xie and Jianing Li and Nazia Bibi and Xiaoxiang Wen and Jianing Li and Sen Yang and Xuegang Lu
},
url = {https://www.nature.com/articles/s41467-025-61967-9.pdf},
doi = {10.1038/s41467-025-61967-9},
year = {2025},
date = {2025-07-18},
journal = {Nature Communications},
volume = {16},
number = {6640},
abstract = {Developing room-temperature phosphorescent (RTP) materials with microscale periodic structures presents a promising prospect for future optical applications but remains challenging due to the complex integration of luminescent and structural components. Herein, we present a strategy for large-scale production of RTP silica nanospheres (RTP SiO2 NPs) with a low dispersity in size using a modified Stöber method, where organic molecules are embedded in silica networks and subsequently undergo in-situ carbonization, aggregation and crystallization to form phosphorescent carbon dots under high-temperature calcination. These NPs can self-assemble into photonic crystal (PC) structures, enabling the straightforward integration of structural color, fluorescence (FL) and RTP to achieve multimodal luminescent properties. The angle-dependent photonic bandgap (PBG) generated by the physical periodic structure modulates light propagation in RTP PC gels, creating FL and RTP angle-dependent chromatic responses. Temperature-induced refractive index changes between SiO2 and the liquid matrix further enable dynamic control of light-scattering states, significantly altering transmittance and emission intensities of FL and RTP. This fusion of physical photonic structures with luminescence offers potential approach for constructing advanced multimodal luminescent devices.},
note = {Room temperature phosphorescent (RTP) materials are promising for optical applications. Here, the authors demonstrate a scalable preparation of RTP nanospheres and their assembly into photonic crystals with multimodal luminescence.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Developing room-temperature phosphorescent (RTP) materials with microscale periodic structures presents a promising prospect for future optical applications but remains challenging due to the complex integration of luminescent and structural components. Herein, we present a strategy for large-scale production of RTP silica nanospheres (RTP SiO2 NPs) with a low dispersity in size using a modified Stöber method, where organic molecules are embedded in silica networks and subsequently undergo in-situ carbonization, aggregation and crystallization to form phosphorescent carbon dots under high-temperature calcination. These NPs can self-assemble into photonic crystal (PC) structures, enabling the straightforward integration of structural color, fluorescence (FL) and RTP to achieve multimodal luminescent properties. The angle-dependent photonic bandgap (PBG) generated by the physical periodic structure modulates light propagation in RTP PC gels, creating FL and RTP angle-dependent chromatic responses. Temperature-induced refractive index changes between SiO2 and the liquid matrix further enable dynamic control of light-scattering states, significantly altering transmittance and emission intensities of FL and RTP. This fusion of physical photonic structures with luminescence offers potential approach for constructing advanced multimodal luminescent devices. |
| 239. | | Xuhua Lan, Lin Geng, Zhen Zhang, Yunfei Li, Jian Yuan, Chen-Xu Zhou, Song Huang, Ziyi Hu, Jing Li, Chengpeng Yang, Yong Zhang, Zhaochuan Fan, Dan Tian, Xiaoxu Zhao, Qingwen Li, Lixing Kang Tunable synthesis of atomic one-dimensional VxTey magnets within single-walled carbon nanotubes In: Nature Communications, vol. 16, no. 6300, 2025, (Unstable configurations and uncontrollable stoichiometries in 1D magnets hinder their practical use. Here, the authors synthesize air-stable 1D VxTey with distinct stoichiometries inside SWCNTs and analyze their magnetic properties.). @article{nokey,
title = {Tunable synthesis of atomic one-dimensional VxTey magnets within single-walled carbon nanotubes},
author = {Xuhua Lan and Lin Geng and Zhen Zhang and Yunfei Li and Jian Yuan and Chen-Xu Zhou and Song Huang and Ziyi Hu and Jing Li and Chengpeng Yang and Yong Zhang and Zhaochuan Fan and Dan Tian and Xiaoxu Zhao and Qingwen Li and Lixing Kang},
url = {https://www.nature.com/articles/s41467-025-61591-7.pdf},
doi = {10.1038/s41467-025-61591-7},
year = {2025},
date = {2025-07-08},
journal = {Nature Communications},
volume = {16},
number = {6300},
abstract = {The unstable configurations and uncontrollable stoichiometric ratios of atomically-thick one-dimensional (1D) magnets pose challenges for practical applications. Here, we employ a spatially confined domain strategy to obtain 1D vanadium tellurides (VxTey) with distinctive stoichiometry within single-walled carbon nanotubes (SWCNTs). Confined by SWCNTs with different inner diameters, three unconventional air-stable VxTey can be generated: 1D 1H-VTe2, V6Te6, and VTe3. Atomically resolved electron microscopy systematically unveils the conformational distributions of these three phases inside SWCNTs. Density functional theory (DFT) calculations indicate that these diverse VxTey phases exhibit different intrinsic electronic structures, which correspond to ferromagnetic, antiferromagnetic, and non-magnetic properties. Furthermore, the magnetic response and magnetic anisotropy of the 1D VxTey@SWCNTs assembly are experimentally confirmed. This work highlights the preparation of air-stable atomic 1D magnets, offering promising solutions for the design of next-generation spintronic devices.},
note = {Unstable configurations and uncontrollable stoichiometries in 1D magnets hinder their practical use. Here, the authors synthesize air-stable 1D VxTey with distinct stoichiometries inside SWCNTs and analyze their magnetic properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The unstable configurations and uncontrollable stoichiometric ratios of atomically-thick one-dimensional (1D) magnets pose challenges for practical applications. Here, we employ a spatially confined domain strategy to obtain 1D vanadium tellurides (VxTey) with distinctive stoichiometry within single-walled carbon nanotubes (SWCNTs). Confined by SWCNTs with different inner diameters, three unconventional air-stable VxTey can be generated: 1D 1H-VTe2, V6Te6, and VTe3. Atomically resolved electron microscopy systematically unveils the conformational distributions of these three phases inside SWCNTs. Density functional theory (DFT) calculations indicate that these diverse VxTey phases exhibit different intrinsic electronic structures, which correspond to ferromagnetic, antiferromagnetic, and non-magnetic properties. Furthermore, the magnetic response and magnetic anisotropy of the 1D VxTey@SWCNTs assembly are experimentally confirmed. This work highlights the preparation of air-stable atomic 1D magnets, offering promising solutions for the design of next-generation spintronic devices. |
| 238. | | Xin Liao, Yun Chen, Tao Xie, Rui-Jing Sun, Lian-Zhi Yang, Chao-Fei Liu, Rui Wang, Svetlana Klyatskaya, Mario Ruben, Wenhao Zhang, Ying-Shuang Fu Spatially dependent f-π exchange interaction within a single-molecule magnet TbPc2 In: Nature Communications, vol. 16, no. 6263, 2025, (Detecting the localized nature of 4f orbitals in lanthanide ion is a major challenge. Here, the authors resolve the spatially dependent f-π exchange interaction within a single-molecule magnet TbPc2 and show their spin states can be electrically controlled in a reversible manner.). @article{nokey,
title = {Spatially dependent f-π exchange interaction within a single-molecule magnet TbPc2},
author = {Xin Liao and Yun Chen and Tao Xie and Rui-Jing Sun and Lian-Zhi Yang and Chao-Fei Liu and Rui Wang and Svetlana Klyatskaya and Mario Ruben and Wenhao Zhang and Ying-Shuang Fu },
url = {https://www.nature.com/articles/s41467-025-61594-4.pdf},
doi = {10.1038/s41467-025-61594-4},
year = {2025},
date = {2025-07-07},
journal = {Nature Communications},
volume = {16},
number = {6263},
abstract = {Electrically probing the spin state of localized f electrons in a rare-earth-based single-molecule magnet, along with understanding its intramolecular magnetic coupling, is of crucial importance for applications in quantum information and advanced spintronics, yet it remains experimentally challenging. Herein, within a single-molecule magnet terbium(III) bis(phthalocyaninato) (TbPc2) double-decker molecule adsorbed on a bilayer graphene epitaxially grown on a SiC(0001) substrate, we experimentally demonstrate a spatially dependent exchange interaction between the magnetic moment of the localized Tb 4f electron and the unpaired spin of the Pc π-radical. The magnetic state of TbPc2, associated with the f-π interaction, is evidently detected through the spectroscopic Kondo resonance and a zero-field Kondo splitting, which can be reversibly switched in a charge/discharge process triggered by the tip-molecule distance. Furthermore, we theoretically describe how the Kondo resonance evolves at the molecular scale, which is mediated by the f-π exchange interaction with its strength varying spatially in a radial decay fashion. Our spatially resolved Kondo characteristics offer a quantitative understanding of the many-body spin correlation, which is coupled with the charge states in a nonuniform and spatially extended system.},
note = {Detecting the localized nature of 4f orbitals in lanthanide ion is a major challenge. Here, the authors resolve the spatially dependent f-π exchange interaction within a single-molecule magnet TbPc2 and show their spin states can be electrically controlled in a reversible manner.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Electrically probing the spin state of localized f electrons in a rare-earth-based single-molecule magnet, along with understanding its intramolecular magnetic coupling, is of crucial importance for applications in quantum information and advanced spintronics, yet it remains experimentally challenging. Herein, within a single-molecule magnet terbium(III) bis(phthalocyaninato) (TbPc2) double-decker molecule adsorbed on a bilayer graphene epitaxially grown on a SiC(0001) substrate, we experimentally demonstrate a spatially dependent exchange interaction between the magnetic moment of the localized Tb 4f electron and the unpaired spin of the Pc π-radical. The magnetic state of TbPc2, associated with the f-π interaction, is evidently detected through the spectroscopic Kondo resonance and a zero-field Kondo splitting, which can be reversibly switched in a charge/discharge process triggered by the tip-molecule distance. Furthermore, we theoretically describe how the Kondo resonance evolves at the molecular scale, which is mediated by the f-π exchange interaction with its strength varying spatially in a radial decay fashion. Our spatially resolved Kondo characteristics offer a quantitative understanding of the many-body spin correlation, which is coupled with the charge states in a nonuniform and spatially extended system. |
| 237. | | Kendra M. Kreienbrink, Zoe A. Cruse, Alisha Kumari, C. Wyatt Shields IV Precise surface patches on active particles of arbitrary shape through microstenciling In: Nature Communications, vol. 16, no. 6062, 2025, (Active particles are useful for microscale functions, but they lack the necessary complexity to be suitable for wide application. Here, the authors present a fabrication method to create patchy active particles of arbitrary shape by microstenciling.). @article{nokey,
title = {Precise surface patches on active particles of arbitrary shape through microstenciling},
author = {Kendra M. Kreienbrink and Zoe A. Cruse and Alisha Kumari and C. Wyatt Shields IV },
url = {https://www.nature.com/articles/s41467-025-61218-x.pdf},
doi = {10.1038/s41467-025-61218-x},
year = {2025},
date = {2025-07-02},
urldate = {2025-07-02},
journal = {Nature Communications},
volume = {16},
number = {6062},
abstract = {Active particles, which locally dissipate energy from their environment to function, are useful across disciplines given their dynamic and programmable behaviors. Altering particle shape or surface asymmetry has led to advancements in controlled locomotion or collective behavior for diverse applications such as microrobotics or biomedicine. However, making arbitrary active particles of precise shape and surface composition remains a significant challenge due to limitations in conventional fabrication methods. This paper introduces a fabrication technique that combines two-photon lithography with sacrificial stencil masking to deposit arbitrary metallic patches onto particles of any shape with a limit of resolution as low as 0.2 µm. Using this method, we demonstrate three varieties of active particles displaying nonconventional dynamics: electrokinetic active spheres with tunable three-dimensional motions, catalytic microdiscs with chiral axial spinning, and steric magnetic particles forming self-limiting microrobots. Overall, this high-resolution microstenciling technique offers a versatile strategy to create well-defined active particles and microrobots for numerous practical uses.},
note = {Active particles are useful for microscale functions, but they lack the necessary complexity to be suitable for wide application. Here, the authors present a fabrication method to create patchy active particles of arbitrary shape by microstenciling.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Active particles, which locally dissipate energy from their environment to function, are useful across disciplines given their dynamic and programmable behaviors. Altering particle shape or surface asymmetry has led to advancements in controlled locomotion or collective behavior for diverse applications such as microrobotics or biomedicine. However, making arbitrary active particles of precise shape and surface composition remains a significant challenge due to limitations in conventional fabrication methods. This paper introduces a fabrication technique that combines two-photon lithography with sacrificial stencil masking to deposit arbitrary metallic patches onto particles of any shape with a limit of resolution as low as 0.2 µm. Using this method, we demonstrate three varieties of active particles displaying nonconventional dynamics: electrokinetic active spheres with tunable three-dimensional motions, catalytic microdiscs with chiral axial spinning, and steric magnetic particles forming self-limiting microrobots. Overall, this high-resolution microstenciling technique offers a versatile strategy to create well-defined active particles and microrobots for numerous practical uses. |
| 236. | | Yuxin Tan, Bin Zhao, Daofu Yuan, Fan Li, Shanshan Yu, Mengda Jin, Yu Zhang, Chang Luo, Wentao Chen, Tao Wang, Jikai Zhu, Ziwei Wang, Tiangang Yang, Shu Liu, Uwe Manthe, Xingan Wang, Dong H. Zhang, Xueming Yang Revealing umbrella bending as a reporter mode in the D+CH4 reaction In: Nature Communications, vol. 16, no. 5893, 2025, (Revealing umbrella bending as a reporter mode in the D+CH4 reaction
The effects of non-reacting components on polyatomic reactions are still largely unclear. Here, the authors show through a combined experimental and theoretical study of the D + CH4 reaction that the CH3 umbrella bending mode serves as a reporter mode, revealing how the D atom dynamically approaches CH4.). @article{nokey,
title = {Revealing umbrella bending as a reporter mode in the D+CH4 reaction},
author = {Yuxin Tan and Bin Zhao and Daofu Yuan and Fan Li and Shanshan Yu and Mengda Jin and Yu Zhang and Chang Luo and Wentao Chen and Tao Wang and Jikai Zhu and Ziwei Wang and Tiangang Yang and Shu Liu and Uwe Manthe and Xingan Wang and Dong H. Zhang and Xueming Yang },
url = {https://www.nature.com/articles/s41467-025-61117-1.pdf},
doi = {10.1038/s41467-025-61117-1},
year = {2025},
date = {2025-07-01},
journal = {Nature Communications},
volume = {16},
number = {5893},
abstract = {How the non-reacting moiety of a molecule influences a polyatomic reaction has been a topic of much research interest. Here we present a comprehensive investigation of the D + CH4 → HD + CH3 reaction, a benchmark polyatomic elementary reaction with CH3 as the non-reacting moiety, employing a high-resolution crossed molecular beams apparatus and an accurate seven-dimensional wave packet method. An interesting angular distribution of the CH3(v’ = 1) product umbrella bending vibrational state is observed to scatter more in the sideways direction than the CH3(v’ = 0) one. By monitoring the wave functions on a dividing surface in the transition state region, the CH3 umbrella bending mode is established as a reporter mode that faithfully reveals how the D atom dynamically approaches CH4 at different total angular momenta or impact parameters. This discovery of the reporter mode provides an opportunity for the detailed study of polyatomic reaction dynamics.},
note = {Revealing umbrella bending as a reporter mode in the D+CH4 reaction
The effects of non-reacting components on polyatomic reactions are still largely unclear. Here, the authors show through a combined experimental and theoretical study of the D + CH4 reaction that the CH3 umbrella bending mode serves as a reporter mode, revealing how the D atom dynamically approaches CH4.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
How the non-reacting moiety of a molecule influences a polyatomic reaction has been a topic of much research interest. Here we present a comprehensive investigation of the D + CH4 → HD + CH3 reaction, a benchmark polyatomic elementary reaction with CH3 as the non-reacting moiety, employing a high-resolution crossed molecular beams apparatus and an accurate seven-dimensional wave packet method. An interesting angular distribution of the CH3(v’ = 1) product umbrella bending vibrational state is observed to scatter more in the sideways direction than the CH3(v’ = 0) one. By monitoring the wave functions on a dividing surface in the transition state region, the CH3 umbrella bending mode is established as a reporter mode that faithfully reveals how the D atom dynamically approaches CH4 at different total angular momenta or impact parameters. This discovery of the reporter mode provides an opportunity for the detailed study of polyatomic reaction dynamics. |
| 235. | | Xiao Meng, Yisheng He, Botyo Dimitrov, Bowen Jin, Zhenghua Zhu, Liang Yuan, Vladimir V. Tsukruk, Chunhong Ye Dynamic mechanical modulation of chiroptical structures via linearly assembled plasmonic nanoparticles on birefringent polymer films In: Nature Communications, vol. 16, no. 5156, 2025, (Plasmonic chiroptical materials suffer from the challenge of combining intense signals with dynamic tunability. Here, the authors present chiroptically active materials composed of linearly assembled gold nanoparticles with birefringent polymer films that enable dynamic tuning of CD signals by mechanical stretching.). @article{nokey,
title = {Dynamic mechanical modulation of chiroptical structures via linearly assembled plasmonic nanoparticles on birefringent polymer films},
author = {Xiao Meng and Yisheng He and Botyo Dimitrov and Bowen Jin and Zhenghua Zhu and Liang Yuan and Vladimir V. Tsukruk and Chunhong Ye },
url = {https://www.nature.com/articles/s41467-025-60165-x.pdf},
doi = {10.1038/s41467-025-60165-x},
year = {2025},
date = {2025-06-03},
journal = {Nature Communications},
volume = {16},
number = {5156},
abstract = {Plasmonic chiral materials exhibiting strong dynamic circular dichroism (CD) with a high asymmetric g-factor are desired for diverse optical applications such as optical communication, especially in near infrared (NIR) spectral range but rarely achieved. Here, we show a chiroptical flexible film composed of an array of linearly aligned plasmonic nanosphere chains with an elastic polymer substrate. These nanomaterials are capable of light polarization rotation up to 40° and a g-factor of 1.71 in NIR wavelength. Such high ellipticity stems from the interplay of high linear dichroism (LD) and linear birefringence (LB). The nanoparticle chains manifest LD at plasmonic resonance, which, combined with the stretching-induced LB of the elastic substrate providing phase retardance, as confirmed by simulation. The configuration offers dynamical and reversible tuning of all critical chiroptical characteristics, including handedness, magnitude, and frequency, via the assembly angle of plasmonic chains, chain length, and the stretching degree of substrates. This strategy is universally applicable to various polymer substrates by combining with plasmonic nanoparticle assembly, enabling the design of flexible chiroptical materials with strong and dynamically tunable chiroptical performance.},
note = {Plasmonic chiroptical materials suffer from the challenge of combining intense signals with dynamic tunability. Here, the authors present chiroptically active materials composed of linearly assembled gold nanoparticles with birefringent polymer films that enable dynamic tuning of CD signals by mechanical stretching.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Plasmonic chiral materials exhibiting strong dynamic circular dichroism (CD) with a high asymmetric g-factor are desired for diverse optical applications such as optical communication, especially in near infrared (NIR) spectral range but rarely achieved. Here, we show a chiroptical flexible film composed of an array of linearly aligned plasmonic nanosphere chains with an elastic polymer substrate. These nanomaterials are capable of light polarization rotation up to 40° and a g-factor of 1.71 in NIR wavelength. Such high ellipticity stems from the interplay of high linear dichroism (LD) and linear birefringence (LB). The nanoparticle chains manifest LD at plasmonic resonance, which, combined with the stretching-induced LB of the elastic substrate providing phase retardance, as confirmed by simulation. The configuration offers dynamical and reversible tuning of all critical chiroptical characteristics, including handedness, magnitude, and frequency, via the assembly angle of plasmonic chains, chain length, and the stretching degree of substrates. This strategy is universally applicable to various polymer substrates by combining with plasmonic nanoparticle assembly, enabling the design of flexible chiroptical materials with strong and dynamically tunable chiroptical performance. |
| 234. | | Finn L. Sebastian, Leon Kaminski, Christoph Bendel, Yohei Yomogida, Yuuya Hosokawa, Han Li, Sebastian Lindenthal, Benjamin S. Flavel, Kazuhiro Yanagi, Jana Zaumseil Circular dichroism of quantum defects in carbon nanotubes created by photocatalytic oxygen functionalization In: Nature Communications, vol. 16, no. 5107, 2025, (Chiral carbon nanotubes interact differently with left- and right-circularly polarized light. Here, the authors use photocatalytic oxygen functionalization of nanotube enantiomers to show that localized quantum defects inherit these chiral properties.). @article{nokey,
title = {Circular dichroism of quantum defects in carbon nanotubes created by photocatalytic oxygen functionalization},
author = {Finn L. Sebastian and Leon Kaminski and Christoph Bendel and Yohei Yomogida and Yuuya Hosokawa and Han Li and Sebastian Lindenthal and Benjamin S. Flavel and Kazuhiro Yanagi and Jana Zaumseil },
url = {https://www.nature.com/articles/s41467-025-60342-y.pdf},
doi = {10.1038/s41467-025-60342-y},
year = {2025},
date = {2025-06-02},
journal = {Nature Communications},
volume = {16},
number = {5107},
abstract = {Control over the chiroptical properties of low-dimensional semiconductors is a promising route toward next-generation optoelectronics and photonics. With their helical chirality, single-wall carbon nanotubes (SWCNTs) offer a suitable framework for exploring chiral excitonic states. In addition to intrinsic, one-dimensional excitons, the targeted functionalization of SWCNTs with luminescent defects introduces zero-dimensional quantum states that enhance photoluminescence quantum yields and exhibit single-photon emission at room temperature. Here, we demonstrate that these defect states inherit the chirality of the respective SWCNT enantiomer, as evident from near-infrared circular dichroism. This observation is achieved by utilizing photocatalysis for efficient and versatile functionalization of SWCNTs with luminescent oxygen defects. The employed approach, based on anthraquinone derivatives as photocatalysts, is applicable to SWCNTs with different diameters, in aqueous or organic dispersions, with different surfactants, and even enables lateral patterning of defects in SWCNT networks. Low catalyst concentrations and the absence of cytotoxic metals or reactants make this functionalization method highly biocompatible. Introducing luminescent defects with uniform binding configurations in sorted nanotube enantiomers represents a key step toward chirality control of quantum defects in SWCNTs.},
note = {Chiral carbon nanotubes interact differently with left- and right-circularly polarized light. Here, the authors use photocatalytic oxygen functionalization of nanotube enantiomers to show that localized quantum defects inherit these chiral properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Control over the chiroptical properties of low-dimensional semiconductors is a promising route toward next-generation optoelectronics and photonics. With their helical chirality, single-wall carbon nanotubes (SWCNTs) offer a suitable framework for exploring chiral excitonic states. In addition to intrinsic, one-dimensional excitons, the targeted functionalization of SWCNTs with luminescent defects introduces zero-dimensional quantum states that enhance photoluminescence quantum yields and exhibit single-photon emission at room temperature. Here, we demonstrate that these defect states inherit the chirality of the respective SWCNT enantiomer, as evident from near-infrared circular dichroism. This observation is achieved by utilizing photocatalysis for efficient and versatile functionalization of SWCNTs with luminescent oxygen defects. The employed approach, based on anthraquinone derivatives as photocatalysts, is applicable to SWCNTs with different diameters, in aqueous or organic dispersions, with different surfactants, and even enables lateral patterning of defects in SWCNT networks. Low catalyst concentrations and the absence of cytotoxic metals or reactants make this functionalization method highly biocompatible. Introducing luminescent defects with uniform binding configurations in sorted nanotube enantiomers represents a key step toward chirality control of quantum defects in SWCNTs. |
| 233. | | Bowen Zhang, Pingda Xu, Jinyun Wang, Zhenyu Hong, Weili Wang, Fuping Dai Overcoming the trade-off between conductivity and strength in copper alloys through undercooling In: Nature Communications, vol. 16, no. 4978, 2025, (Copper alloys with high strength and conductivity are essential for modern industries. Here, the authors find an approach to enhance the conductivity of Cu-Be alloys by inducing spherical Be-rich clusters through deep undercooling.). @article{nokey,
title = {Overcoming the trade-off between conductivity and strength in copper alloys through undercooling},
author = {Bowen Zhang and Pingda Xu and Jinyun Wang and Zhenyu Hong and Weili Wang and Fuping Dai },
url = {https://www.nature.com/articles/s41467-025-60346-8.pdf},
doi = {10.1038/s41467-025-60346-8},
year = {2025},
date = {2025-05-29},
journal = {Nature Communications},
volume = {16},
number = {4978},
abstract = {With the continuous development of high-performance copper alloys in modern industries, it becomes increasingly challenging to further enhance their conductivities. The key bottleneck is the existence of an upper limit on the amount of precipitation, leading to inadequate purification of the copper matrix. Here we demonstrate a phenomenon of significant conductivity enhancement in a Cu-Be alloy through undercooling. It shows that lots of spherical Be-rich clusters can spontaneously form in the deeply undercooled alloy. These clusters survive after subsequent solution treatment and are independent from the normal precipitates during aging, thereby leading to additional purification of the copper matrix. Under peak aging, the electrical conductivity of the undercooled alloy reaches up to 80% International Annealed Cu Standard, which is 30% higher than that of the same component alloy prepared in a conventional way, while its strength remains high. Our study provides an alternative way to address the long-standing strength-conductivity trade-off in copper alloys.},
note = {Copper alloys with high strength and conductivity are essential for modern industries. Here, the authors find an approach to enhance the conductivity of Cu-Be alloys by inducing spherical Be-rich clusters through deep undercooling.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
With the continuous development of high-performance copper alloys in modern industries, it becomes increasingly challenging to further enhance their conductivities. The key bottleneck is the existence of an upper limit on the amount of precipitation, leading to inadequate purification of the copper matrix. Here we demonstrate a phenomenon of significant conductivity enhancement in a Cu-Be alloy through undercooling. It shows that lots of spherical Be-rich clusters can spontaneously form in the deeply undercooled alloy. These clusters survive after subsequent solution treatment and are independent from the normal precipitates during aging, thereby leading to additional purification of the copper matrix. Under peak aging, the electrical conductivity of the undercooled alloy reaches up to 80% International Annealed Cu Standard, which is 30% higher than that of the same component alloy prepared in a conventional way, while its strength remains high. Our study provides an alternative way to address the long-standing strength-conductivity trade-off in copper alloys. |
| 232. | | Yangjian Cai, Ming Lu, Xian Qin, Dayong Jin, Jiajia Zhou Understanding shell coating effects to overcome quenching in single anisotropic upconversion nanoparticles In: Nature Communications, vol. 16, no. 4927, 2025, (Shell coatings reduce luminescence quenching in spherical nanoparticles for upconversion, but anisotropic nanoparticles are less well studied. Here, the authors investigate the effects of coatings on individual nanorods and show that their effectiveness depends on power density and shell types.). @article{nokey,
title = {Understanding shell coating effects to overcome quenching in single anisotropic upconversion nanoparticles},
author = {Yangjian Cai and Ming Lu and Xian Qin and Dayong Jin and Jiajia Zhou },
url = {https://www.nature.com/articles/s41467-025-60347-7.pdf},
doi = {10.1038/s41467-025-60347-7},
year = {2025},
date = {2025-05-27},
journal = {Nature Communications},
volume = {16},
number = {4927},
abstract = {Shell coating is known to suppress luminescence quenching in spherical upconversion nanoparticles. However, the emergence of anisotropic nanoparticles with facet-selective, directional growth complicates the coating process, and the use of traditional active, inert, or polymer coatings on such structures remains largely unexplored. Here, we synthesize a series of nanorods with designed geometries, enabling quantitative spectral analysis at the single-particle level. We observe that directional growth of inert or active shells at the rod tips enhances emission relative to the parent core, with their relative effectiveness governed by power density and shell thickness. Ligand presence—polymer or oleate—quenches upconversion relative to bare nanorods. Although local heating is observed at the single-particle level, it does not affect spectroscopic observations, ligand stability, or data reproducibility. Our findings reveal how directionally grown shells influence the optical properties of upconversion nanorods, providing essential insights for their future applications in bioimaging, sensing, and photonics.},
note = {Shell coatings reduce luminescence quenching in spherical nanoparticles for upconversion, but anisotropic nanoparticles are less well studied. Here, the authors investigate the effects of coatings on individual nanorods and show that their effectiveness depends on power density and shell types.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Shell coating is known to suppress luminescence quenching in spherical upconversion nanoparticles. However, the emergence of anisotropic nanoparticles with facet-selective, directional growth complicates the coating process, and the use of traditional active, inert, or polymer coatings on such structures remains largely unexplored. Here, we synthesize a series of nanorods with designed geometries, enabling quantitative spectral analysis at the single-particle level. We observe that directional growth of inert or active shells at the rod tips enhances emission relative to the parent core, with their relative effectiveness governed by power density and shell thickness. Ligand presence—polymer or oleate—quenches upconversion relative to bare nanorods. Although local heating is observed at the single-particle level, it does not affect spectroscopic observations, ligand stability, or data reproducibility. Our findings reveal how directionally grown shells influence the optical properties of upconversion nanorods, providing essential insights for their future applications in bioimaging, sensing, and photonics. |
| 231. | | Aisheng Song, Jian-Xun Zhao, Xin Tang, Hai-Jun Wu, Zhiyue Xu, Jiawei Cao, Xiao Liu, Hui Wang, Qunyang Li, Yuan-Zhong Hu, Xin Li, Jianbin Luo, Tian-Bao Ma Tuning friction force and reducing wear by applying alternating electric current in conductive AFM experiments In: Nature Communications, vol. 16, no. 4704, 2025, (Friction and wear cause widespread energy loss and damage. Here, the authors propose a strategy to reduce friction and wear through inducing dynamic electronic density redistribution via alternating electric current.). @article{nokey,
title = {Tuning friction force and reducing wear by applying alternating electric current in conductive AFM experiments},
author = {Aisheng Song and Jian-Xun Zhao and Xin Tang and Hai-Jun Wu and Zhiyue Xu and Jiawei Cao and Xiao Liu and Hui Wang and Qunyang Li and Yuan-Zhong Hu and Xin Li and Jianbin Luo and Tian-Bao Ma},
url = {https://www.nature.com/articles/s41467-025-59989-4.pdf},
doi = {10.1038/s41467-025-59989-4},
year = {2025},
date = {2025-05-20},
journal = {Nature Communications},
volume = {16},
number = {4704},
abstract = {Reducing friction has been a human pursuit for centuries, and is especially important for the development of nanotechnology. Nowadays, with the atomic-level understanding of friction, it is possible to reduce friction by modulating the configuration and motion of interfacial atoms. However, how to further reduce friction by modulating the interfacial electronic properties is still unclear. Here we show a strategy to achieve friction and wear reduction through inducing dynamic electronic density redistribution via alternating electric current. The friction force between conductive Ir AFM tip and graphene on Ni substrate can be reduced to 1/4 under 1 kHz alternating current, and maintain for more than 70,000 s under 9.1 GPa contact pressure without any obvious wear. An electronic-level friction model (PTT-E model) is presented to unravel and quantify the tuning effect, showing that the alternating current induced dynamic electron density redistribution is the key to friction reduction. This work proposes a feasible and robust method to reduce friction and wear in nanomechanical devices, and advances the understanding and predicting of electronic contribution in friction tuning.},
note = {Friction and wear cause widespread energy loss and damage. Here, the authors propose a strategy to reduce friction and wear through inducing dynamic electronic density redistribution via alternating electric current.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Reducing friction has been a human pursuit for centuries, and is especially important for the development of nanotechnology. Nowadays, with the atomic-level understanding of friction, it is possible to reduce friction by modulating the configuration and motion of interfacial atoms. However, how to further reduce friction by modulating the interfacial electronic properties is still unclear. Here we show a strategy to achieve friction and wear reduction through inducing dynamic electronic density redistribution via alternating electric current. The friction force between conductive Ir AFM tip and graphene on Ni substrate can be reduced to 1/4 under 1 kHz alternating current, and maintain for more than 70,000 s under 9.1 GPa contact pressure without any obvious wear. An electronic-level friction model (PTT-E model) is presented to unravel and quantify the tuning effect, showing that the alternating current induced dynamic electron density redistribution is the key to friction reduction. This work proposes a feasible and robust method to reduce friction and wear in nanomechanical devices, and advances the understanding and predicting of electronic contribution in friction tuning. |
| 230. | | Aleksandr Bashkatov, Florian Bürkle, Çayan Demirkırv, Wei Ding, Vatsal Sanjay, Alexander Babich, Xuegeng Yang, Gerd Mutschke, Jürgen Czarske, Detlef Lohse, Dominik Krug, Lars Büttner, Kerstin Eckert Electrolyte droplet spraying in H2 bubbles during water electrolysis under normal and microgravity conditions In: Nature Communications, vol. 16, no. 4580, 2025, (Electrogenerated gas bubbles impair the efficiency of electrolysis and require a deeper insight into their dynamics. Here, the authors describe a transport mechanism in which electrolyte droplets from the coalescence of microbubbles are sprayed into H2 bubbles.). @article{nokey,
title = {Electrolyte droplet spraying in H2 bubbles during water electrolysis under normal and microgravity conditions},
author = {Aleksandr Bashkatov and Florian Bürkle and Çayan Demirkırv and Wei Ding and Vatsal Sanjay and Alexander Babich and Xuegeng Yang and Gerd Mutschke and Jürgen Czarske and Detlef Lohse and Dominik Krug and Lars Büttner and Kerstin Eckert },
url = {https://www.nature.com/articles/s41467-025-59762-7.pdf},
doi = {10.1038/s41467-025-59762-7},
year = {2025},
date = {2025-05-16},
journal = {Nature Communications},
volume = {16},
number = {4580},
abstract = {Electrolytically generated gas bubbles can significantly hamper the overall electrolysis efficiency. Therefore it is crucial to understand their dynamics in order to optimise water electrolyzer systems. Herein, we elucidate a distinct transport mechanism whereby electrolyte droplets are sprayed into H2 bubbles. These droplets arise from the fragmentation of the Worthington jet, which is engendered by the coalescence with microbubbles. The robustness of this phenomenon is corroborated under both normal and microgravity conditions. Reminiscent of bursting bubbles on a liquid-gas interface, electrolyte spraying results in a flow inside the bubble. This flow couples, in an intriguing way, with the thermocapillary convection at the bubble’s surface, clearly underlining the high interfacial mobility. In the case of electrode-attached bubbles, the sprayed droplets form electrolyte puddles affecting the dynamics near the three-phase contact line and favoring bubble detachment from the electrode. The results of this work unravel important insights into the physico-chemical aspects of electrolytic gas bubbles, integral for optimizing gas-evolving electrochemical systems.},
note = {Electrogenerated gas bubbles impair the efficiency of electrolysis and require a deeper insight into their dynamics. Here, the authors describe a transport mechanism in which electrolyte droplets from the coalescence of microbubbles are sprayed into H2 bubbles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Electrolytically generated gas bubbles can significantly hamper the overall electrolysis efficiency. Therefore it is crucial to understand their dynamics in order to optimise water electrolyzer systems. Herein, we elucidate a distinct transport mechanism whereby electrolyte droplets are sprayed into H2 bubbles. These droplets arise from the fragmentation of the Worthington jet, which is engendered by the coalescence with microbubbles. The robustness of this phenomenon is corroborated under both normal and microgravity conditions. Reminiscent of bursting bubbles on a liquid-gas interface, electrolyte spraying results in a flow inside the bubble. This flow couples, in an intriguing way, with the thermocapillary convection at the bubble’s surface, clearly underlining the high interfacial mobility. In the case of electrode-attached bubbles, the sprayed droplets form electrolyte puddles affecting the dynamics near the three-phase contact line and favoring bubble detachment from the electrode. The results of this work unravel important insights into the physico-chemical aspects of electrolytic gas bubbles, integral for optimizing gas-evolving electrochemical systems. |
| 229. | | William Trewby, Mahdi Tavakol, Kislon Voïtchovsky Local mapping of the nanoscale viscoelastic properties of fluid membranes by AFM nanorheology In: Nature Communications, vol. 16, no. 3842 , 2025, (The viscoelasticity of fluid membranes is challenging to quantify with nanometre precision. Here, the authors develop an AFM-based nanorheology platform to measure local molecular mobilities and spatial correlations in lateral diffusion within the membrane.). @article{nokey,
title = {Local mapping of the nanoscale viscoelastic properties of fluid membranes by AFM nanorheology},
author = {William Trewby and Mahdi Tavakol and Kislon Voïtchovsky },
url = {https://www.nature.com/articles/s41467-025-59260-w.pdf},
doi = {10.1038/s41467-025-59260-w},
year = {2025},
date = {2025-04-24},
journal = {Nature Communications},
volume = {16},
number = {3842 },
abstract = {Biological membranes are intrinsically dynamic entities that continually adapt their biophysical properties and molecular organisation to support cellular function. Current microscopy techniques can derive high-resolution structural information of labelled molecules but quantifying the associated viscoelastic behaviour with nanometre precision remains challenging. Here, we develop an approach based on atomic force microscopy in conjunction with fast nano-actuators to map the viscoelastic response of unlabelled supported membranes with nanometre spatial resolution. On fluid membranes, we show that the method can quantify local variations in the molecular mobility of the lipids and derive a diffusion coefficient. We confirm our experimental approach with molecular dynamics simulations, also highlighting the role played by the water at the interface with the membrane on the measurement. Probing ternary model bilayers reveals spatial correlations in the local diffusion over distances of ≈20 nm within liquid disordered domains. This lateral correlation is enhanced in native bovine lens membranes, where the inclusion of protein-rich domains induces four-fold variations in the diffusion coefficient across <100 nm of the fluid regions, consistent with biological function. Our findings suggest that diffusion is highly localised in fluid biomembranes.},
note = {The viscoelasticity of fluid membranes is challenging to quantify with nanometre precision. Here, the authors develop an AFM-based nanorheology platform to measure local molecular mobilities and spatial correlations in lateral diffusion within the membrane.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Biological membranes are intrinsically dynamic entities that continually adapt their biophysical properties and molecular organisation to support cellular function. Current microscopy techniques can derive high-resolution structural information of labelled molecules but quantifying the associated viscoelastic behaviour with nanometre precision remains challenging. Here, we develop an approach based on atomic force microscopy in conjunction with fast nano-actuators to map the viscoelastic response of unlabelled supported membranes with nanometre spatial resolution. On fluid membranes, we show that the method can quantify local variations in the molecular mobility of the lipids and derive a diffusion coefficient. We confirm our experimental approach with molecular dynamics simulations, also highlighting the role played by the water at the interface with the membrane on the measurement. Probing ternary model bilayers reveals spatial correlations in the local diffusion over distances of ≈20 nm within liquid disordered domains. This lateral correlation is enhanced in native bovine lens membranes, where the inclusion of protein-rich domains induces four-fold variations in the diffusion coefficient across <100 nm of the fluid regions, consistent with biological function. Our findings suggest that diffusion is highly localised in fluid biomembranes. |
| 228. | | Zhongzheng Yu, Wen Kiat Chan, Donglei Zhou, Xinjuan Li, Yang Lu, Zhao Jiang, Bofeng Xue, Huangtianzhi Zhu, Simon Dowland, Junzhi Ye, Alasdair Tew, Lars van Turnhout, Qichun Gu, Linjie Dai, Tianjun Liu, Caterina Ducati, Akshay Rao, Timothy Thatt Yang Tan Overcoming lattice mismatch for core-shell NaGdF4@CsPbBr3 heterostructures In: Nature Communications, vol. 16, no. 3891, 2025, (Lattice mismatches are a difficult obstacle in the formation of core-shell heterostructures. Here, the authors develop a strategy to overcome the lattice mismatch and grow α-phase lead halide perovskites onto β-phase lanthanide-doped nanoparticles.). @article{nokey,
title = {Overcoming lattice mismatch for core-shell NaGdF4@CsPbBr3 heterostructures},
author = {Zhongzheng Yu and Wen Kiat Chan and Donglei Zhou and Xinjuan Li and Yang Lu and Zhao Jiang and Bofeng Xue and Huangtianzhi Zhu and Simon Dowland and Junzhi Ye and Alasdair Tew and Lars van Turnhout and Qichun Gu and Linjie Dai and Tianjun Liu and Caterina Ducati and Akshay Rao and Timothy Thatt Yang Tan },
url = {https://www.nature.com/articles/s41467-025-59315-y.pdf},
doi = {10.1038/s41467-025-59315-y},
year = {2025},
date = {2025-04-24},
journal = {Nature Communications},
volume = {16},
number = {3891},
abstract = {The formation of core-shell heterostructures allows direct contact of two components for more efficient energy transfer while requires exquisite lattice match. Lattice mismatch is one of the most challenging obstacles for combining two components with different phases. In this work, we develop a strategy to overcome the limitation of lattice mismatch and grow α-phase lead halide perovskites (LHPs) onto β-phase lanthanide-doped nanoparticles (LnNPs) by seeding sub-8 nm LnNPs. This LnNP@LHP heterostructure effectively passivates the surface defects of LnNPs to obtain enhanced upconversion performance and enables two-way energy transfer within the heterostructures. We identify and prove that core size along with a high reaction temperature, instead of phase, is critical to overcome the lattice mismatch. Our strategy uncovers insights into the key factor of direct growth for heterostructures and we believe the current synthesis strategy for high-quality heterostructures will have strong application potential in optoelectronics, anticounterfeiting and light detection.},
note = {Lattice mismatches are a difficult obstacle in the formation of core-shell heterostructures. Here, the authors develop a strategy to overcome the lattice mismatch and grow α-phase lead halide perovskites onto β-phase lanthanide-doped nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The formation of core-shell heterostructures allows direct contact of two components for more efficient energy transfer while requires exquisite lattice match. Lattice mismatch is one of the most challenging obstacles for combining two components with different phases. In this work, we develop a strategy to overcome the limitation of lattice mismatch and grow α-phase lead halide perovskites (LHPs) onto β-phase lanthanide-doped nanoparticles (LnNPs) by seeding sub-8 nm LnNPs. This LnNP@LHP heterostructure effectively passivates the surface defects of LnNPs to obtain enhanced upconversion performance and enables two-way energy transfer within the heterostructures. We identify and prove that core size along with a high reaction temperature, instead of phase, is critical to overcome the lattice mismatch. Our strategy uncovers insights into the key factor of direct growth for heterostructures and we believe the current synthesis strategy for high-quality heterostructures will have strong application potential in optoelectronics, anticounterfeiting and light detection. |
| 227. | | Dunzhu Li, Peijing Li, Yunhong Shi, Emmet D. Sheerin, Zihan Zhang, Luming Yang, Liwen Xiao, Christopher Hill, Conall Gordon, Manuel Ruether, Joshua Pepper, John E. Sader, Michael A. Morris, Jing Jing Wang, John J. Boland Stress-induced phase separation in plastics drives the release of amorphous polymer micropollutants into water In: Nature Communications, vol. 16, no. 3814, 2025, (Residual stresses are pervasive in most modern plastics. Here, the authors show that they promote the migration of low-molecular-weight amorphous polymer and additives from bulk plastics and release micropollutants into water.). @article{nokey,
title = {Stress-induced phase separation in plastics drives the release of amorphous polymer micropollutants into water},
author = {Dunzhu Li and Peijing Li and Yunhong Shi and Emmet D. Sheerin and Zihan Zhang and Luming Yang and Liwen Xiao and Christopher Hill and Conall Gordon and Manuel Ruether and Joshua Pepper and John E. Sader and Michael A. Morris and Jing Jing Wang and John J. Boland },
url = {https://www.nature.com/articles/s41467-025-58898-w.pdf},
doi = {10.1038/s41467-025-58898-w},
year = {2025},
date = {2025-04-23},
journal = {Nature Communications},
volume = {16},
number = {3814},
abstract = {Residual stress is an intrinsic property of semicrystalline plastics such as polypropylene and polyethylene. However, there is no fundamental understanding of the role intrinsic residual stress plays in the generation of plastic pollutants that threaten the environment and human health. Here, we show that the processing-induced compressive residual stress typically found in polypropylene and polyethylene plastics forces internal nano and microscale segregation of low molecular weight (MW) amorphous polymer droplets onto the plastic’s surface. Squeeze flow simulations reveal this stress-driven volumetric flow is consistent with that of a Bingham plastic material, with a temperature-dependent threshold yield stress. We confirm that flow is thermally activated and stress dependent, with a reduced energy barrier at higher compressive stresses. Transfer of surface segregated droplets into water generates amorphous polymer micropollutants (APMPs) that are denatured, with structure and composition different from that of traditional polycrystalline microplastics. Studies with water-containing plastic bottles show that the highly compressed bottle neck and mouth regions are predominantly responsible for the release of APMPs. Our findings reveal a stress-induced mechanism of plastic degradation and underscore the need to modify current plastic processing technologies to reduce residual stress levels and suppress phase separation of low MW APMPs in plastics.},
note = {Residual stresses are pervasive in most modern plastics. Here, the authors show that they promote the migration of low-molecular-weight amorphous polymer and additives from bulk plastics and release micropollutants into water.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Residual stress is an intrinsic property of semicrystalline plastics such as polypropylene and polyethylene. However, there is no fundamental understanding of the role intrinsic residual stress plays in the generation of plastic pollutants that threaten the environment and human health. Here, we show that the processing-induced compressive residual stress typically found in polypropylene and polyethylene plastics forces internal nano and microscale segregation of low molecular weight (MW) amorphous polymer droplets onto the plastic’s surface. Squeeze flow simulations reveal this stress-driven volumetric flow is consistent with that of a Bingham plastic material, with a temperature-dependent threshold yield stress. We confirm that flow is thermally activated and stress dependent, with a reduced energy barrier at higher compressive stresses. Transfer of surface segregated droplets into water generates amorphous polymer micropollutants (APMPs) that are denatured, with structure and composition different from that of traditional polycrystalline microplastics. Studies with water-containing plastic bottles show that the highly compressed bottle neck and mouth regions are predominantly responsible for the release of APMPs. Our findings reveal a stress-induced mechanism of plastic degradation and underscore the need to modify current plastic processing technologies to reduce residual stress levels and suppress phase separation of low MW APMPs in plastics. |
| 226. | | Linfeng Xu, Zetan Cao, Zhiwen Liu, Cheng Zheng, Simin Peng, Yong Lu, Haoran Liu, Bin Chen Filming evolution dynamics of Hg nanodroplets mediated at solid-gas and solid-liquid interfaces by in-situ TEM In: Nature Communications, vol. 16, no. 3684, 2025, (The in-situ visualization of the evolution of metal nanodroplets at multiphase interfaces remains a challenge. Here, the authors directly observe the motion and jetting behaviors of Hg nanodroplets regulated at solid-gas and solid-liquid interfaces.). @article{nokey,
title = {Filming evolution dynamics of Hg nanodroplets mediated at solid-gas and solid-liquid interfaces by in-situ TEM},
author = {Linfeng Xu and Zetan Cao and Zhiwen Liu and Cheng Zheng and Simin Peng and Yong Lu and Haoran Liu and Bin Chen },
url = {https://www.nature.com/articles/s41467-025-59063-z.pdf},
doi = {10.1038/s41467-025-59063-z},
year = {2025},
date = {2025-04-17},
journal = {Nature Communications},
volume = {16},
number = {3684},
abstract = {Nanodroplets at multiphase interfaces are ubiquitous in nature with implications ranging from fundamental interfacial science to industrial applications including catalytic, environmental, biological and medical processes. Direct observation of full dynamic evolutions of liquid metal nanodroplets at nanoscale multiphase interfaces offers indispensable insights, however, remains challenging and unclear. Here, we fabricate gas and liquid cells containing HgS nanocrystals through electrospinning and achieve the statistical investigations of full picture of Hg nanodroplets evolving at solid-gas and solid-liquid interfaces by in-situ transmission electron microscopy. In the gas cells, the voids nucleate, grow and coalesce into the crack-like feature along the <001> direction, while Hg nanodroplets form, move rapidly on the ratchet surface and are evolved into bigger ones through the nanobridges. Distinctly, mediated by the solid-liquid interface, the liquid Hg with the ink-like feature jets in the liquid cells. Such ink-jetting behavior occurs multiple times with the intervals from several to several tens of seconds, which is modulated through the competition between reductive electrons and oxidative species derived from the radiolysis of liquids. In-depth understanding of distinct nanodroplets dynamics at nanoscale solid-gas and solid-liquid interfaces offers a feasible approach for designing liquid metal-based nanocomplexes with regulatory interfacial, morphological and rheological functionalities.},
note = {The in-situ visualization of the evolution of metal nanodroplets at multiphase interfaces remains a challenge. Here, the authors directly observe the motion and jetting behaviors of Hg nanodroplets regulated at solid-gas and solid-liquid interfaces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanodroplets at multiphase interfaces are ubiquitous in nature with implications ranging from fundamental interfacial science to industrial applications including catalytic, environmental, biological and medical processes. Direct observation of full dynamic evolutions of liquid metal nanodroplets at nanoscale multiphase interfaces offers indispensable insights, however, remains challenging and unclear. Here, we fabricate gas and liquid cells containing HgS nanocrystals through electrospinning and achieve the statistical investigations of full picture of Hg nanodroplets evolving at solid-gas and solid-liquid interfaces by in-situ transmission electron microscopy. In the gas cells, the voids nucleate, grow and coalesce into the crack-like feature along the <001> direction, while Hg nanodroplets form, move rapidly on the ratchet surface and are evolved into bigger ones through the nanobridges. Distinctly, mediated by the solid-liquid interface, the liquid Hg with the ink-like feature jets in the liquid cells. Such ink-jetting behavior occurs multiple times with the intervals from several to several tens of seconds, which is modulated through the competition between reductive electrons and oxidative species derived from the radiolysis of liquids. In-depth understanding of distinct nanodroplets dynamics at nanoscale solid-gas and solid-liquid interfaces offers a feasible approach for designing liquid metal-based nanocomplexes with regulatory interfacial, morphological and rheological functionalities. |
| 225. | | Xiaona Zhao, Xiao-Li Zhou, Cheng-Xin Cao, Xin Xi, Xian-Wei Liu Plasmonic in-situ imaging of zeta potential distributions at electrochemical interfaces of 2D materials in water In: Nature Communications, vol. 16, no. 3494, 2025, (Understanding the electrical double layer at solid-liquid interfaces is crucial for various technologies. Here, the authors present an optical method for direct visualization and quantification of the zeta potential at 2D material interfaces.). @article{nokey,
title = {Plasmonic in-situ imaging of zeta potential distributions at electrochemical interfaces of 2D materials in water},
author = {Xiaona Zhao and Xiao-Li Zhou and Cheng-Xin Cao and Xin Xi and Xian-Wei Liu },
url = {https://www.nature.com/articles/s41467-025-58793-4.pdf},
doi = {10.1038/s41467-025-58793-4},
year = {2025},
date = {2025-04-12},
journal = {Nature Communications},
volume = {16},
number = {3494},
abstract = {Understanding the electrical double layer (EDL) at solid-liquid interfaces is pivotal across various fields, including energy storage, electrowetting, and electrocatalysis, yet probing its structure and heterogeneity remains a considerable challenge. Here, we report an optical method for the direct visualization and quantification of the zeta potential (ζ) across the interfaces between 2D materials and aqueous solutions. By modulating surface charge density, we map the heterogenous distribution of ζ potential across the MoS2 nanosheet interface, revealing how both external factors and intrinsic material properties shape interfacial charge. This approach overcomes the drawbacks of conventional methods for evaluating ζ potential in 2D materials, providing insights into elucidate the complex interplay between the ζ potential and the catalytic activity of 2D materials. Furthermore, it establishes a robust framework for exploring the EDL in various electrochemical systems. Our findings reveal a deeper understanding of complex electrochemical interface interactions, offering valuable insights into the fundamental processes governing these systems.},
note = {Understanding the electrical double layer at solid-liquid interfaces is crucial for various technologies. Here, the authors present an optical method for direct visualization and quantification of the zeta potential at 2D material interfaces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Understanding the electrical double layer (EDL) at solid-liquid interfaces is pivotal across various fields, including energy storage, electrowetting, and electrocatalysis, yet probing its structure and heterogeneity remains a considerable challenge. Here, we report an optical method for the direct visualization and quantification of the zeta potential (ζ) across the interfaces between 2D materials and aqueous solutions. By modulating surface charge density, we map the heterogenous distribution of ζ potential across the MoS2 nanosheet interface, revealing how both external factors and intrinsic material properties shape interfacial charge. This approach overcomes the drawbacks of conventional methods for evaluating ζ potential in 2D materials, providing insights into elucidate the complex interplay between the ζ potential and the catalytic activity of 2D materials. Furthermore, it establishes a robust framework for exploring the EDL in various electrochemical systems. Our findings reveal a deeper understanding of complex electrochemical interface interactions, offering valuable insights into the fundamental processes governing these systems. |
| 224. | | Shuangshuang Zeng, Tian Tian, Jiwoo Oh, Zhan-Hong Lin, Chih-Jen Shih Direct nanopatterning of complex 3D surfaces and self-aligned superlattices via molecular-beam holographic lithography In: Nature Communications, vol. 16, no. 3436, 2025, (Conventional lithography methods involve the transfer of patterns through resist templating. Here, the authors explore a concept based on the Moiré interference of molecular beams to directly write complex 3D surfaces and self-aligned superlattices.). @article{nokey,
title = {Direct nanopatterning of complex 3D surfaces and self-aligned superlattices via molecular-beam holographic lithography},
author = {Shuangshuang Zeng and Tian Tian and Jiwoo Oh and Zhan-Hong Lin and Chih-Jen Shih },
url = {https://www.nature.com/articles/s41467-025-58651-3.pdf},
doi = {10.1038/s41467-025-58651-3},
year = {2025},
date = {2025-04-11},
journal = {Nature Communications},
volume = {16},
number = {3436},
abstract = {Conventional lithography methods involving pattern transfer through resist templating face challenges of material compatibility with various process solvents. Other approaches of direct material writing often compromise pattern complexity and overlay accuracy. Here we explore a concept based on the Moiré interference of molecular beams to directly pattern complex three-dimensional (3D) surfaces made by any evaporable materials, such as metals, oxides and organic semiconductors. Our proposed approach, termed the molecular-beam holographic lithography (MBHL), relies on precise control over angular projections of material flux passing through nanoapertures superimposed on the substrate, emulating the interference of coherent laser beams in interference lithography. Incorporating with our computational lithography (CL) algorithm, we have demonstrated self-aligned overlay of multiple material patterns to yield binary up to quinary superlattices, with a critical dimension and overlay accuracy on the order of 50 and 2 nm, respectively. The process is expected to substantially expand the boundary of materials combination for high-throughput fabrication of complex superstructures of translational symmetry on arbitrary substrates, enabling emerging nanoimaging, sensing, catalysis, and optoelectronic devices.},
note = {Conventional lithography methods involve the transfer of patterns through resist templating. Here, the authors explore a concept based on the Moiré interference of molecular beams to directly write complex 3D surfaces and self-aligned superlattices.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Conventional lithography methods involving pattern transfer through resist templating face challenges of material compatibility with various process solvents. Other approaches of direct material writing often compromise pattern complexity and overlay accuracy. Here we explore a concept based on the Moiré interference of molecular beams to directly pattern complex three-dimensional (3D) surfaces made by any evaporable materials, such as metals, oxides and organic semiconductors. Our proposed approach, termed the molecular-beam holographic lithography (MBHL), relies on precise control over angular projections of material flux passing through nanoapertures superimposed on the substrate, emulating the interference of coherent laser beams in interference lithography. Incorporating with our computational lithography (CL) algorithm, we have demonstrated self-aligned overlay of multiple material patterns to yield binary up to quinary superlattices, with a critical dimension and overlay accuracy on the order of 50 and 2 nm, respectively. The process is expected to substantially expand the boundary of materials combination for high-throughput fabrication of complex superstructures of translational symmetry on arbitrary substrates, enabling emerging nanoimaging, sensing, catalysis, and optoelectronic devices. |
| 223. | | Youhua Lu, Ye-Guang Fang, Yang Chen, Han Xue, Junqiang Mao, Bo Guan, Jie Liu, Jinping Li, Libo Li, Chongqin Zhu, Wei-Hai Fang, Thomas P. Russell, Jianjun Wang Sandwiching of MOF nanoparticles between graphene oxide nanosheets among ice grains In: Nature Communications, vol. 16, no. 3397, 2025, (Conventional methods for nanoparticle cluster formation involve surface functionalization, which can alter the properties. Here, the authors propose a non-disruptive strategy to position nanomaterials of different shapes between nanosheets without the need for pre-modification.). @article{nokey,
title = {Sandwiching of MOF nanoparticles between graphene oxide nanosheets among ice grains},
author = {Youhua Lu and Ye-Guang Fang and Yang Chen and Han Xue and Junqiang Mao and Bo Guan and Jie Liu and Jinping Li and Libo Li and Chongqin Zhu and Wei-Hai Fang and Thomas P. Russell and Jianjun Wang },
url = {https://www.nature.com/articles/s41467-025-56949-w.pdf},
doi = {10.1038/s41467-025-56949-w},
year = {2025},
date = {2025-04-10},
journal = {Nature Communications},
volume = {16},
number = {3397},
abstract = {Current strategies to tailor the formation of nanoparticle clusters require specificity and directionality built into the surface functionalization of the nanoparticles by involved chemistries that can alter their properties. Here, we describe a non-disruptive approach to place nanomaterials of different shapes between nanosheets, i.e., nano-sandwiches, absent any pre-modification of the components. We demonstrate this with metal-organic frameworks (MOFs) and silicon oxide (SiO2) nanoparticles sandwiched between graphene oxide (GO) nanosheets, MOF-GO and SiO2-GO, respectively. For the MOF-GO, the MOF shows significantly enhanced conductivity and retains its original crystallinity, even after one-year exposure to aqueous acid/base solutions, where the GO effectively encapsulates the MOF, shielding it from polar molecules and ions. The MOF-GOs are shown to effectively capture CO2 from a high-humidity flue gas while fully maintaining their crystallinities and porosities. Similar behavior is found for other MOFs, including water-sensitive HKUST-1 and MOF-5, promoting the use of MOFs in practical applications. The nanoparticle sandwich strategy provides opportunities for materials science in the design of nanoparticle clusters consisting of different materials and shapes with predetermined spatial arrangements.},
note = {Conventional methods for nanoparticle cluster formation involve surface functionalization, which can alter the properties. Here, the authors propose a non-disruptive strategy to position nanomaterials of different shapes between nanosheets without the need for pre-modification.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Current strategies to tailor the formation of nanoparticle clusters require specificity and directionality built into the surface functionalization of the nanoparticles by involved chemistries that can alter their properties. Here, we describe a non-disruptive approach to place nanomaterials of different shapes between nanosheets, i.e., nano-sandwiches, absent any pre-modification of the components. We demonstrate this with metal-organic frameworks (MOFs) and silicon oxide (SiO2) nanoparticles sandwiched between graphene oxide (GO) nanosheets, MOF-GO and SiO2-GO, respectively. For the MOF-GO, the MOF shows significantly enhanced conductivity and retains its original crystallinity, even after one-year exposure to aqueous acid/base solutions, where the GO effectively encapsulates the MOF, shielding it from polar molecules and ions. The MOF-GOs are shown to effectively capture CO2 from a high-humidity flue gas while fully maintaining their crystallinities and porosities. Similar behavior is found for other MOFs, including water-sensitive HKUST-1 and MOF-5, promoting the use of MOFs in practical applications. The nanoparticle sandwich strategy provides opportunities for materials science in the design of nanoparticle clusters consisting of different materials and shapes with predetermined spatial arrangements. |
| 222. | | Sida Wang, Rowan Walker-Gibbons, Bethany Watkins, Binghui Lin, Madhavi Krishnan
Chemical control of colloidal self-assembly driven by the electrosolvation force In: Nature Communications, vol. 16, no. 2872, 2025, (The electrosolvation force drives a counterintuitive attraction between particles with the same electrical charge in solution. Here the authors show that the force can be modulated by molecular species that are known to alter the interfacial water structure.). @article{nokey,
title = {Chemical control of colloidal self-assembly driven by the electrosolvation force},
author = {Sida Wang and Rowan Walker-Gibbons and Bethany Watkins and Binghui Lin and Madhavi Krishnan
},
url = {https://www.nature.com/articles/s41467-025-57953-w.pdf},
doi = {10.1038/s41467-025-57953-w},
year = {2025},
date = {2025-03-24},
urldate = {2025-03-24},
journal = {Nature Communications},
volume = {16},
number = {2872},
abstract = {Self-assembly of matter in solution generally relies on attractive interactions that overcome entropy and drive the formation of higher-order molecular and particulate structures. Such interactions are central to a variety of molecular processes, e.g., crystallisation, biomolecular folding and condensation, pathological protein aggregation and biofouling. The electrosolvation force introduces a distinct conceptual paradigm to the existing palette of interactions that govern the spontaneous accretion and organisation of matter. However, an understanding of the underlying physical chemistry, and therefore the ability to exert control over and tune the interaction, remains incomplete. Here we provide further evidence that this force arises from the structure of the interfacial electrolyte. Neutral molecules such as a different solvent, osmolytes or surfactants, may — even at very low concentrations in the medium — disrupt or reinforce pre-existing interfacial solvent structure, thereby delivering unanticipated chemical tuning of the ability of matter to self-assemble. The observations present unexpected mechanistic elements that may explain the impact of co-solvents and osmolytes on protein structure, stability and biomolecular condensation. Our findings thus furnish insight into the microscopic mechanisms that drive the emergence of order and structure from molecular to macroscopic scales in the solution phase.},
note = {The electrosolvation force drives a counterintuitive attraction between particles with the same electrical charge in solution. Here the authors show that the force can be modulated by molecular species that are known to alter the interfacial water structure.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Self-assembly of matter in solution generally relies on attractive interactions that overcome entropy and drive the formation of higher-order molecular and particulate structures. Such interactions are central to a variety of molecular processes, e.g., crystallisation, biomolecular folding and condensation, pathological protein aggregation and biofouling. The electrosolvation force introduces a distinct conceptual paradigm to the existing palette of interactions that govern the spontaneous accretion and organisation of matter. However, an understanding of the underlying physical chemistry, and therefore the ability to exert control over and tune the interaction, remains incomplete. Here we provide further evidence that this force arises from the structure of the interfacial electrolyte. Neutral molecules such as a different solvent, osmolytes or surfactants, may — even at very low concentrations in the medium — disrupt or reinforce pre-existing interfacial solvent structure, thereby delivering unanticipated chemical tuning of the ability of matter to self-assemble. The observations present unexpected mechanistic elements that may explain the impact of co-solvents and osmolytes on protein structure, stability and biomolecular condensation. Our findings thus furnish insight into the microscopic mechanisms that drive the emergence of order and structure from molecular to macroscopic scales in the solution phase. |
| 221. | | Jeongwon Kim, Qiang Zhao, Inyoung Choi, Myeong Jin Oh, Sunwoo Kwon, Sungho Park Ensemble hot-spots in 3D supercrystals of plasmonic octahedral nanoparticles in tip-to-tip configured superlattices In: Nature Communications, vol. 16, no. 2762, 2025, (Nanoparticle arrangement in a tip-to-tip configuration is challenging. Here, the authors report a synthetic method for organizing octahedral gold nanoparticles in a distinct upright 3D superstructure where the pointed tips are aligned with neighboring nanoparticles.). @article{nokey,
title = {Ensemble hot-spots in 3D supercrystals of plasmonic octahedral nanoparticles in tip-to-tip configured superlattices},
author = {Jeongwon Kim and Qiang Zhao and Inyoung Choi and Myeong Jin Oh and Sunwoo Kwon and Sungho Park },
url = {https://www.nature.com/articles/s41467-025-58029-5.pdf},
doi = {10.1038/s41467-025-58029-5},
year = {2025},
date = {2025-03-20},
journal = {Nature Communications},
volume = {16},
number = {2762},
abstract = {Nanoparticle assembly offers promising strategy for harnessing the physicochemical interparticle interactions. Despite its potential for boosting light-matter interaction, achieving nanoparticle assembly with tip-to-tip manner remains a significant challenge. Here we show a synthetic procedure for organizing gold octahedral nanoparticles into a distinct three-dimensional upright superstructure, where the pointed tips are oriented toward neighboring nanoparticles to promote enhanced near-field focusing at these apexes. This arrangement, referred to as the “coupling of the lightning rod effect”, facilitates production in the form of “superpowder”, which exhibits an extensive assembly order like a powder. Deviating from natural packing principles, this tip-to-tip alignment—the upright octahedral superlattice—optimizes near-field focusing on its vertices while maintaining consistently high porosity, allowing for deep penetration of adsorbates. This configuration is advantageous for enabling surface-enhanced Raman scattering of gaseous molecules with reduced background fluorescence signals, particularly under high-intensity laser excitation, a challenging feat with conventional surface-enhanced Raman scattering techniques.},
note = {Nanoparticle arrangement in a tip-to-tip configuration is challenging. Here, the authors report a synthetic method for organizing octahedral gold nanoparticles in a distinct upright 3D superstructure where the pointed tips are aligned with neighboring nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanoparticle assembly offers promising strategy for harnessing the physicochemical interparticle interactions. Despite its potential for boosting light-matter interaction, achieving nanoparticle assembly with tip-to-tip manner remains a significant challenge. Here we show a synthetic procedure for organizing gold octahedral nanoparticles into a distinct three-dimensional upright superstructure, where the pointed tips are oriented toward neighboring nanoparticles to promote enhanced near-field focusing at these apexes. This arrangement, referred to as the “coupling of the lightning rod effect”, facilitates production in the form of “superpowder”, which exhibits an extensive assembly order like a powder. Deviating from natural packing principles, this tip-to-tip alignment—the upright octahedral superlattice—optimizes near-field focusing on its vertices while maintaining consistently high porosity, allowing for deep penetration of adsorbates. This configuration is advantageous for enabling surface-enhanced Raman scattering of gaseous molecules with reduced background fluorescence signals, particularly under high-intensity laser excitation, a challenging feat with conventional surface-enhanced Raman scattering techniques. |
| 220. | | Aritra Biswas, Nir Lemcoff, Ofir Shelonchik, Mark Baranov, Gil Gordon, Uri Ben Nun, Yossi Weizmann Molecular light-to-heat conversion promotes orthogonal synthesis and assembly of metal-organic frameworks In: Nature Communications, vol. 16, no. 2758, 2025, (Controlling the reaction temperature is critical in chemistry. Here, the authors harness molecular-photothermal conversion to synthesize and assemble MOFs with tunable structures under different light wavelengths for advanced materials.). @article{nokey,
title = {Molecular light-to-heat conversion promotes orthogonal synthesis and assembly of metal-organic frameworks},
author = {Aritra Biswas and Nir Lemcoff and Ofir Shelonchik and Mark Baranov and Gil Gordon and Uri Ben Nun and Yossi Weizmann },
url = {https://www.nature.com/articles/s41467-025-57933-0.pdf},
doi = {10.1038/s41467-025-57933-0},
year = {2025},
date = {2025-03-20},
journal = {Nature Communications},
volume = {16},
number = {2758},
abstract = {Temperature is a fundamental parameter in any chemical process, affecting reaction rates, selectivity and more. In this regard, photon-assisted heat generation for chemical reactions utilizing photothermal materials is emerging as an exciting tool for innovative research. Herein, we develop a synthesis and in-situ assembly strategy for metal-organic frameworks (MOFs) based on the distinct heating of photothermal materials under visible light. A simple cobalt chloride molecular complex is utilized as an efficient and stable light-to-heat converter for initial MOF formation. A thorough investigation of the assembly mechanism reveals the key role photothermal conversion has in the synthesis of the superstructures. Finally, palladium nanoparticles (PdNPs) are utilized as competing photothermal agents (PTAs) shedding light on the dynamics between different heat sources within a reaction and resulting in MOF-NP composites. This work highlights the versatility of the photothermal approach in the synthesis of advanced materials introducing a promising route to the micro/nano assembly of different materials.},
note = {Controlling the reaction temperature is critical in chemistry. Here, the authors harness molecular-photothermal conversion to synthesize and assemble MOFs with tunable structures under different light wavelengths for advanced materials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Temperature is a fundamental parameter in any chemical process, affecting reaction rates, selectivity and more. In this regard, photon-assisted heat generation for chemical reactions utilizing photothermal materials is emerging as an exciting tool for innovative research. Herein, we develop a synthesis and in-situ assembly strategy for metal-organic frameworks (MOFs) based on the distinct heating of photothermal materials under visible light. A simple cobalt chloride molecular complex is utilized as an efficient and stable light-to-heat converter for initial MOF formation. A thorough investigation of the assembly mechanism reveals the key role photothermal conversion has in the synthesis of the superstructures. Finally, palladium nanoparticles (PdNPs) are utilized as competing photothermal agents (PTAs) shedding light on the dynamics between different heat sources within a reaction and resulting in MOF-NP composites. This work highlights the versatility of the photothermal approach in the synthesis of advanced materials introducing a promising route to the micro/nano assembly of different materials. |
| 219. | | Xiao Wang, Zhi Qiao, Zhu Fang, Yufeng Zhai, Runze Yu, Gang Chen In-situ X-ray scattering observation of colloidal epitaxy at the gas-liquid-solid interface In: Nature Communications, vol. 16, no. 2687, 2025, (Convective self-assembly of particles is widely employed, but some fundamental aspects remain unknown. Here, the authors perform an in-situ SAXS study during the convective self-assembly of colloidal particles at the meniscus, which provides mechanistic insights.). @article{nokey,
title = {In-situ X-ray scattering observation of colloidal epitaxy at the gas-liquid-solid interface},
author = {Xiao Wang and Zhi Qiao and Zhu Fang and Yufeng Zhai and Runze Yu and Gang Chen },
url = {https://www.nature.com/articles/s41467-025-58028-6.pdf},
doi = {10.1038/s41467-025-58028-6},
year = {2025},
date = {2025-03-19},
urldate = {2025-03-19},
journal = {Nature Communications},
volume = {16},
number = {2687},
abstract = {The convective self-assembly of dip-coating is a long-established technique widely employed in scientific and industrial applications. Despite its apparent importance, many of the fundamental aspects remain unknown, particularly the exact assembling mechanism and its relationship with evaporation kinetics and fluid dynamics. Here, we perform the in-situ small-angle X-ray scattering study of the real-time convective self-assembly of colloidal particles inside a meniscus. This approach allows us to resolve the transient assembling processes occurring at the gas, liquid and solid interfaces. Together with ex-situ scanning electron microscopy measurements via the freeze-dry method, the colloidal epitaxy process is uncovered, where the multilayer is sequentially assembled using the interfacial monolayer as a template. The microscopic ordering of the final multilayer is highly correlated with that of the initial monolayer. The evaporation kinetics and fluid dynamics are numerically simulated, which rationalizes the monolayer formation and the dynamic epitaxial process.},
note = {Convective self-assembly of particles is widely employed, but some fundamental aspects remain unknown. Here, the authors perform an in-situ SAXS study during the convective self-assembly of colloidal particles at the meniscus, which provides mechanistic insights.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The convective self-assembly of dip-coating is a long-established technique widely employed in scientific and industrial applications. Despite its apparent importance, many of the fundamental aspects remain unknown, particularly the exact assembling mechanism and its relationship with evaporation kinetics and fluid dynamics. Here, we perform the in-situ small-angle X-ray scattering study of the real-time convective self-assembly of colloidal particles inside a meniscus. This approach allows us to resolve the transient assembling processes occurring at the gas, liquid and solid interfaces. Together with ex-situ scanning electron microscopy measurements via the freeze-dry method, the colloidal epitaxy process is uncovered, where the multilayer is sequentially assembled using the interfacial monolayer as a template. The microscopic ordering of the final multilayer is highly correlated with that of the initial monolayer. The evaporation kinetics and fluid dynamics are numerically simulated, which rationalizes the monolayer formation and the dynamic epitaxial process. |
| 218. | | Xiaoyu Song, Yahui Li, Meng Yin, Junfang Li, Haifeng Yang, Wei Liu, Xiaotian Wang, Guangcheng Xi Multilayered hollow transition metal nitride spheres made from single-source precursors for SERS analytics In: Nature Communications, vol. 16, no. 2678, 2025, (Harsh reaction pathways make transition metal nitride (TMN) grains susceptible to sintering. Here, the authors report a general route to synthesize multilayered TMN hollow spheres with high specific surface area from a single-source precursor.). @article{nokey,
title = {Multilayered hollow transition metal nitride spheres made from single-source precursors for SERS analytics},
author = {Xiaoyu Song and Yahui Li and Meng Yin and Junfang Li and Haifeng Yang and Wei Liu and Xiaotian Wang and Guangcheng Xi },
url = {https://www.nature.com/articles/s41467-025-58031-x.pdf},
doi = {10.1038/s41467-025-58031-x},
year = {2025},
date = {2025-03-18},
journal = {Nature Communications},
volume = {16},
number = {2678},
abstract = {Traditional high-temperature and high-pressure synthesis routes make transition metal nitride (TMN) grains prone to sintering and agglomeration, thus synthesis of architectures with high specific surface area and pore volume is an urgent problem to be solved for the applications of TMNs. Here, a general single-source precursor route is designed to synthesize cubic-phase γ-Mo2N multilayered hollow spheres with high specific surface area (191.3 m2 g–1) and pore volume (0.69 cm3 g–1) under relatively mild conditions. Furthermore, by changing the metal composition of the precursor through ion exchange, a series of TMN (WN, TiN, VN, NbN, MoN/WN, MoN/WN/TiN) multilayer hollow spheres with high specific surface area (178.6–193.7 m2 g–1) and pore volume (0.57–0.72 cm3 g–1) are prepared. Particle size of precursor is found to be a key factor affecting the crystal phase and composition of molybdenum nitride nanostructures, and hexagonal-phase δ-MoN hierarchical hollow spheres composed of nanosheets are synthesized by adjusting the precursor particle size. The γ-Mo2N multilayered hollow spheres exhibit enhanced Raman activity for applications in trace detection of polychlorophenol and microplastics.},
note = {Harsh reaction pathways make transition metal nitride (TMN) grains susceptible to sintering. Here, the authors report a general route to synthesize multilayered TMN hollow spheres with high specific surface area from a single-source precursor.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Traditional high-temperature and high-pressure synthesis routes make transition metal nitride (TMN) grains prone to sintering and agglomeration, thus synthesis of architectures with high specific surface area and pore volume is an urgent problem to be solved for the applications of TMNs. Here, a general single-source precursor route is designed to synthesize cubic-phase γ-Mo2N multilayered hollow spheres with high specific surface area (191.3 m2 g–1) and pore volume (0.69 cm3 g–1) under relatively mild conditions. Furthermore, by changing the metal composition of the precursor through ion exchange, a series of TMN (WN, TiN, VN, NbN, MoN/WN, MoN/WN/TiN) multilayer hollow spheres with high specific surface area (178.6–193.7 m2 g–1) and pore volume (0.57–0.72 cm3 g–1) are prepared. Particle size of precursor is found to be a key factor affecting the crystal phase and composition of molybdenum nitride nanostructures, and hexagonal-phase δ-MoN hierarchical hollow spheres composed of nanosheets are synthesized by adjusting the precursor particle size. The γ-Mo2N multilayered hollow spheres exhibit enhanced Raman activity for applications in trace detection of polychlorophenol and microplastics. |
| 217. | | Sebastian Schmitt, Hans Hasse, Simon Stephan Entropy scaling for diffusion coefficients in fluid mixtures In: Nature Communications, vol. 16, no. 2611, 2025, (The consistent description of diffusion coefficients (self-diffusion and mutual diffusion) in liquid mixtures across different phases is a challenge. Here, the authors develop a model based on entropy scaling and molecular-based equation of state.). @article{nokey,
title = {Entropy scaling for diffusion coefficients in fluid mixtures},
author = {Sebastian Schmitt and Hans Hasse and Simon Stephan},
url = {https://www.nature.com/articles/s41467-025-57780-z.pdf},
doi = {10.1038/s41467-025-57780-z},
year = {2025},
date = {2025-03-17},
journal = {Nature Communications},
volume = {16},
number = {2611},
abstract = {Entropy scaling is a powerful technique that has been used for predicting transport properties of pure components over a wide range of states. However, modeling mixture diffusion coefficients by entropy scaling is an unresolved task. We tackle this issue and present an entropy scaling framework for predicting mixture self-diffusion coefficients as well as mutual diffusion coefficients in a thermodynamically consistent way. The predictions of the mixture diffusion coefficients are made based on information on the self-diffusion coefficients of the pure components and the infinite-dilution diffusion coefficients. This is accomplished using information on the entropy of the mixture, which is taken here from molecular-based equations of state. Examples for the application of the entropy scaling framework for the prediction of diffusion coefficients in mixtures illustrate its performance. It enables predictions over a wide range of temperatures and pressures including gaseous, liquid, supercritical, and metastable states—also for strongly non-ideal mixtures.},
note = {The consistent description of diffusion coefficients (self-diffusion and mutual diffusion) in liquid mixtures across different phases is a challenge. Here, the authors develop a model based on entropy scaling and molecular-based equation of state.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Entropy scaling is a powerful technique that has been used for predicting transport properties of pure components over a wide range of states. However, modeling mixture diffusion coefficients by entropy scaling is an unresolved task. We tackle this issue and present an entropy scaling framework for predicting mixture self-diffusion coefficients as well as mutual diffusion coefficients in a thermodynamically consistent way. The predictions of the mixture diffusion coefficients are made based on information on the self-diffusion coefficients of the pure components and the infinite-dilution diffusion coefficients. This is accomplished using information on the entropy of the mixture, which is taken here from molecular-based equations of state. Examples for the application of the entropy scaling framework for the prediction of diffusion coefficients in mixtures illustrate its performance. It enables predictions over a wide range of temperatures and pressures including gaseous, liquid, supercritical, and metastable states—also for strongly non-ideal mixtures. |
| 216. | | Xiaoqian Mi, Lixue Liu, Shujia Yang, Peiqi Wu, Weiqing Zhan, Xinyi Ji, Jiajie Liang
Ink formulation of functional nanowires with hyperbranched stabilizers for versatile printing of flexible electronics In: Nature Communications, vol. 16, no. 2590, 2025, (Depositing flexible electronics based on nanowires uniformly and reproducibly is a challenge. Here, the authors demonstrate the formulation of silver nanowire suspensions using hyperbranched molecules that enable scalable fabrication of electrodes with different printing technologies.). @article{nokey,
title = {Ink formulation of functional nanowires with hyperbranched stabilizers for versatile printing of flexible electronics},
author = {Xiaoqian Mi and Lixue Liu and Shujia Yang and Peiqi Wu and Weiqing Zhan and Xinyi Ji and Jiajie Liang
},
url = {https://www.nature.com/articles/s41467-025-57959-4.pdf},
doi = {10.1038/s41467-025-57959-4},
year = {2025},
date = {2025-03-16},
journal = {Nature Communications},
volume = {16},
number = {2590},
abstract = {Functional nanowire ink formulations require elaborate control over their composition, rheological properties, and fluidic properties to optimize their printing processes. They also require harsh post-fabrication treatments to maximize the performance of the resulting printed flexible devices, making it challenging to uniformly deposit nanowire-based architectures and ensure device reproducibility and scalability. Here, we propose a strategy for developing silver nanowire (AgNW) ink formulations, where hyperbranched molecules (HPMs) are employed as both dispersant and stabilizer for nanowires. The three-dimensional architecture with functional groups on the periphery of HPMs enables the preparation of thixotropic HPMs-AgNW inks with solid contents of up to 20 wt.% in both aqueous and organic solvents using a low amount of HPMs (AgNW and HPMs weight ratio = 1:0.001). The HPMs-AgNW inks can be printed into patterns with a resolution of 20 μm on various flexible substrates without needing harsh post-treatments. We obtain bar-coated transparent electrodes (sheet resistance of 17.1 Ω sq−1 at 94.7% transmittance), slot-die-coated flexible conductive patterns, screen-printed conductive lines (conductivity exceeding 6.2 × 104 S cm−1), and 3D printed stretchable wires. Importantly, this HPMs-stabilized formulation strategy is general for various functional nanowires, enabling the integration of a diverse set of nanowire-based wearable electronic systems.},
note = {Depositing flexible electronics based on nanowires uniformly and reproducibly is a challenge. Here, the authors demonstrate the formulation of silver nanowire suspensions using hyperbranched molecules that enable scalable fabrication of electrodes with different printing technologies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Functional nanowire ink formulations require elaborate control over their composition, rheological properties, and fluidic properties to optimize their printing processes. They also require harsh post-fabrication treatments to maximize the performance of the resulting printed flexible devices, making it challenging to uniformly deposit nanowire-based architectures and ensure device reproducibility and scalability. Here, we propose a strategy for developing silver nanowire (AgNW) ink formulations, where hyperbranched molecules (HPMs) are employed as both dispersant and stabilizer for nanowires. The three-dimensional architecture with functional groups on the periphery of HPMs enables the preparation of thixotropic HPMs-AgNW inks with solid contents of up to 20 wt.% in both aqueous and organic solvents using a low amount of HPMs (AgNW and HPMs weight ratio = 1:0.001). The HPMs-AgNW inks can be printed into patterns with a resolution of 20 μm on various flexible substrates without needing harsh post-treatments. We obtain bar-coated transparent electrodes (sheet resistance of 17.1 Ω sq−1 at 94.7% transmittance), slot-die-coated flexible conductive patterns, screen-printed conductive lines (conductivity exceeding 6.2 × 104 S cm−1), and 3D printed stretchable wires. Importantly, this HPMs-stabilized formulation strategy is general for various functional nanowires, enabling the integration of a diverse set of nanowire-based wearable electronic systems. |
| 215. | | Ujjwal Verma, Isabella M. Riley, Bratislav Lukić, Ludovic Broche, Pieter Verboven, Jan A. Delcour, Bart M. Nicolaï High-speed computed tomography to visualise the 3D microstructural dynamics of oil uptake in deep-fried foods In: Nature Communications, vol. 16, no. 2600, 2025, (Deep-frying produces foods with a porous texture, but this also raises concerns about the final oil content. Here, the authors study the pore formation of dough during rapid heating and show how oil uptake depends on the porous structure created.). @article{nokey,
title = {High-speed computed tomography to visualise the 3D microstructural dynamics of oil uptake in deep-fried foods},
author = {Ujjwal Verma and Isabella M. Riley and Bratislav Lukić and Ludovic Broche and Pieter Verboven and Jan A. Delcour and Bart M. Nicolaï },
url = {https://www.nature.com/articles/s41467-025-57934-z.pdf},
doi = {10.1038/s41467-025-57934-z},
year = {2025},
date = {2025-03-16},
journal = {Nature Communications},
volume = {16},
number = {2600},
abstract = {Oil serves as both the high-temperature heating medium during deep-frying unit operations and a contributor to the organoleptic properties of deep-fried foods. Its absorption is linked to structural deformations during deep-frying that create pathways for oil to enter the food microstructure. This study proposes a 4D imaging system (three spatial dimensions and time) using fast synchrotron radiation tomography for in-situ visualisation during deep-frying and post-frying cooling to understand the mechanisms behind oil absorption. Using wheat flour dough as a model food system, we investigate the impact of frying oil temperature on structural deformation and pore formation in the crust and core, pore structure integrity, and oil uptake and distribution. The results show that higher temperatures lead to the formation of a distinguished crust with surface openings, facilitating greater oil absorption in small crust pores through capillary action. Comparing 3D microstructures attained at different frying oil temperatures, final oil content reaches 14.4% at 180 °C, 12.2% at 150 °C, and 1.3% at 120 °C, with temperature-dependent structural changes in pore connectivity and network integrity significantly impacting the rate of oil uptake and its distribution.},
note = {Deep-frying produces foods with a porous texture, but this also raises concerns about the final oil content. Here, the authors study the pore formation of dough during rapid heating and show how oil uptake depends on the porous structure created.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oil serves as both the high-temperature heating medium during deep-frying unit operations and a contributor to the organoleptic properties of deep-fried foods. Its absorption is linked to structural deformations during deep-frying that create pathways for oil to enter the food microstructure. This study proposes a 4D imaging system (three spatial dimensions and time) using fast synchrotron radiation tomography for in-situ visualisation during deep-frying and post-frying cooling to understand the mechanisms behind oil absorption. Using wheat flour dough as a model food system, we investigate the impact of frying oil temperature on structural deformation and pore formation in the crust and core, pore structure integrity, and oil uptake and distribution. The results show that higher temperatures lead to the formation of a distinguished crust with surface openings, facilitating greater oil absorption in small crust pores through capillary action. Comparing 3D microstructures attained at different frying oil temperatures, final oil content reaches 14.4% at 180 °C, 12.2% at 150 °C, and 1.3% at 120 °C, with temperature-dependent structural changes in pore connectivity and network integrity significantly impacting the rate of oil uptake and its distribution. |
| 214. | | Xiaoxi Luan, Yu Tian, Fengxia Wu, Lu Cheng, Minghua Tang, Xiali Lv, Haili Wei, Xiaodan Wang, Fenghua Li, Guobao Xu, Wenxin Niu Enantioselective synthesis of chiroplasmonic helicoidal nanoparticles by nanoconfinement in chiral dielectric shells In: Nature Communications, vol. 16, no. 2418, 2025, (Strategies for the synthesis of chiral metal nanoparticles were mainly limited to gold. Here, the authors present the enantioselective synthesis of chiroplasmonic helicoidal nanoparticles beyond gold using chiral silica shells.). @article{nokey,
title = {Enantioselective synthesis of chiroplasmonic helicoidal nanoparticles by nanoconfinement in chiral dielectric shells},
author = {Xiaoxi Luan and Yu Tian and Fengxia Wu and Lu Cheng and Minghua Tang and Xiali Lv and Haili Wei and Xiaodan Wang and Fenghua Li and Guobao Xu and Wenxin Niu },
url = {https://www.nature.com/articles/s41467-025-57624-w.pdf},
doi = {10.1038/s41467-025-57624-w},
year = {2025},
date = {2025-03-12},
journal = {Nature Communications},
volume = {16},
number = {2418},
abstract = {Helicoid metal nanoparticles with intrinsic chirality have unveiled tailorable properties and unlocked many chirality-related applications across various fields. Nevertheless, the existing strategies for enantioselective synthesis of helicoid metal nanoparticles have been predominantly limited to gold. Here, we demonstrate a robust and versatile strategy for the enantioselective synthesis of helicoid nanoparticles beyond gold, leveraging chiral nanoconfinement provided by chiral SiO2 or nanoshells. The chiral nanoconfinement strategy enables the decoupling of ligand-directed crystal growth from chiral induction, allowing for the independent tuning of these two critical aspects. As a result, this approach can not only facilitate the replication of chiral shapes from the chiral nanoshells but also allow the generation of alternative chiral shapes. By employing this approach, we demonstrate the enantioselective synthesis of helicoid Pt, Au@Pt, Au@Pd, Au@Ag, and Au@Cu nanoparticles. The chiroplasmonic properties of Pt- and Pd-based chiral nanoparticles have been discovered, and the inversion of chiroplasmonic properties of Ag-based chiral nanoparticles via facet control has been documented and theoretically explained. The chiral nanoconfinement strategy enriches the toolbox for creating chiral nanoparticles and supports their exploration in diverse applications.},
note = {Strategies for the synthesis of chiral metal nanoparticles were mainly limited to gold. Here, the authors present the enantioselective synthesis of chiroplasmonic helicoidal nanoparticles beyond gold using chiral silica shells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Helicoid metal nanoparticles with intrinsic chirality have unveiled tailorable properties and unlocked many chirality-related applications across various fields. Nevertheless, the existing strategies for enantioselective synthesis of helicoid metal nanoparticles have been predominantly limited to gold. Here, we demonstrate a robust and versatile strategy for the enantioselective synthesis of helicoid nanoparticles beyond gold, leveraging chiral nanoconfinement provided by chiral SiO2 or nanoshells. The chiral nanoconfinement strategy enables the decoupling of ligand-directed crystal growth from chiral induction, allowing for the independent tuning of these two critical aspects. As a result, this approach can not only facilitate the replication of chiral shapes from the chiral nanoshells but also allow the generation of alternative chiral shapes. By employing this approach, we demonstrate the enantioselective synthesis of helicoid Pt, Au@Pt, Au@Pd, Au@Ag, and Au@Cu nanoparticles. The chiroplasmonic properties of Pt- and Pd-based chiral nanoparticles have been discovered, and the inversion of chiroplasmonic properties of Ag-based chiral nanoparticles via facet control has been documented and theoretically explained. The chiral nanoconfinement strategy enriches the toolbox for creating chiral nanoparticles and supports their exploration in diverse applications. |
| 213. | | Mengmeng Ma, Qinnan Yu, Jiayi Zhang, Xiao Chen, Wenbo Li, Xianlin Qu, Xuliang Zhang, Jiale Feng, Fei Wei, Jianyu Yuan, Tao Cheng, Sheng Dai, Yi Wang, Bin Song, Boyuan Shen Imaging molecular structures and interactions by enhanced confinement effect in electron microscopy In: Nature Communications, vol. 16, no. 2447, 2025, (Molecular interactions are of great importance in materials science. Here, the authors use the confinement effect and low-dose imaging to reveal the confined molecules and evaluate the strength of host-guest interactions in different material systems.). @article{nokey,
title = {Imaging molecular structures and interactions by enhanced confinement effect in electron microscopy},
author = {Mengmeng Ma and Qinnan Yu and Jiayi Zhang and Xiao Chen and Wenbo Li and Xianlin Qu and Xuliang Zhang and Jiale Feng and Fei Wei and Jianyu Yuan and Tao Cheng and Sheng Dai and Yi Wang and Bin Song and Boyuan Shen },
url = {https://www.nature.com/articles/s41467-025-57816-4.pdf},
doi = {10.1038/s41467-025-57816-4},
year = {2025},
date = {2025-03-12},
journal = {Nature Communications},
volume = {16},
number = {2447},
abstract = {Atomic imaging of molecules and intermolecular interactions are of great significance for a deeper understanding of the basic physics and chemistry in various applications, but it is still challenging in electron microscopy due to their thermal mobility and beam sensitivity. Confinement effect and low-dose imaging method may efficiently help us achieve stable high-resolution resolving of molecules and their interactions. Here, we propose a general strategy to image the confined molecules and evaluate the strengths of host-guest interactions in three material systems by low-dose electron microscopy. Then, we change the guest molecules to analyze how each kind of interaction strength influences the imaging quality of these molecules by using a same parameter, the aspect ratios of imaged molecular projections. In the material systems of perovskites (ionic) and zeolites with adsorbed molecules (van der Waals), we can obtain a clear image of molecular configurations by enhancing host-guest interactions. Even in metal organic framework (coordination) system, the atomic structures and bonds of aromatics can be achieved. These results provide a general description on the relation between molecular images and interactions, making it possible to study more molecular behaviors in wide applications by real-space imaging.},
note = {Molecular interactions are of great importance in materials science. Here, the authors use the confinement effect and low-dose imaging to reveal the confined molecules and evaluate the strength of host-guest interactions in different material systems.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Atomic imaging of molecules and intermolecular interactions are of great significance for a deeper understanding of the basic physics and chemistry in various applications, but it is still challenging in electron microscopy due to their thermal mobility and beam sensitivity. Confinement effect and low-dose imaging method may efficiently help us achieve stable high-resolution resolving of molecules and their interactions. Here, we propose a general strategy to image the confined molecules and evaluate the strengths of host-guest interactions in three material systems by low-dose electron microscopy. Then, we change the guest molecules to analyze how each kind of interaction strength influences the imaging quality of these molecules by using a same parameter, the aspect ratios of imaged molecular projections. In the material systems of perovskites (ionic) and zeolites with adsorbed molecules (van der Waals), we can obtain a clear image of molecular configurations by enhancing host-guest interactions. Even in metal organic framework (coordination) system, the atomic structures and bonds of aromatics can be achieved. These results provide a general description on the relation between molecular images and interactions, making it possible to study more molecular behaviors in wide applications by real-space imaging. |
| 212. | | Bi-Jun Geng, Yun-Fei Li, Lin-Long Deng, Tong-Ruo Diao, Zi-Wei Ma, Long-Xing Lin, Jin-Shu Li, Yi-Dong Yan, Jia-Wei Yan, Yuan-Zhi Tan, Bing-Wei Mao, Su-Yuan Xie, Zhong-Qun Tian, Yang Yang Post-modulation of layer-by-layer assemblies coordinated by a catalytic dose of fullerene derivatives without external fields In: Nature Communications, vol. 16, no. 2276, 2025, (Molecular assembly is of great importance in many fields. Here, the authors establish a strategy to post-modulate angle-mismatched layer-by-layer materials by fullerene derivatives and achieve a modulation accuracy of up to 5 Å.). @article{nokey,
title = {Post-modulation of layer-by-layer assemblies coordinated by a catalytic dose of fullerene derivatives without external fields},
author = {Bi-Jun Geng and Yun-Fei Li and Lin-Long Deng and Tong-Ruo Diao and Zi-Wei Ma and Long-Xing Lin and Jin-Shu Li and Yi-Dong Yan and Jia-Wei Yan and Yuan-Zhi Tan and Bing-Wei Mao and Su-Yuan Xie and Zhong-Qun Tian and Yang Yang },
url = {https://www.nature.com/articles/s41467-025-57626-8.pdf},
doi = {10.1038/s41467-025-57626-8},
year = {2025},
date = {2025-03-07},
journal = {Nature Communications},
volume = {16},
number = {2276},
abstract = {Molecular assembly has attracted wide attention in chemistry, condensed physics, molecular electronics, and materials sciences. However, it remains a great challenge to perform post-modulation on the assembled structures without the aid of externally applied fields. Herein, we demonstrate a combined full-weak-bonded interaction and fullerene-derivative strategy and achieve the post-modulation of layer-by-layer assembly with a precision of 5.0 Å. In the absence of external fields, the fullerene derivative exhibits a long-range interaction to boost the modulation of the assembly. Benefiting from that, a catalytic dose of fullerene derivative is able to modulate a large area of assembly, reminiscent of the catalyst in chemical reactions. This work provides an efficient and flexible approach for the catalysis and precise modulation of molecular assembly.},
note = {Molecular assembly is of great importance in many fields. Here, the authors establish a strategy to post-modulate angle-mismatched layer-by-layer materials by fullerene derivatives and achieve a modulation accuracy of up to 5 Å.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Molecular assembly has attracted wide attention in chemistry, condensed physics, molecular electronics, and materials sciences. However, it remains a great challenge to perform post-modulation on the assembled structures without the aid of externally applied fields. Herein, we demonstrate a combined full-weak-bonded interaction and fullerene-derivative strategy and achieve the post-modulation of layer-by-layer assembly with a precision of 5.0 Å. In the absence of external fields, the fullerene derivative exhibits a long-range interaction to boost the modulation of the assembly. Benefiting from that, a catalytic dose of fullerene derivative is able to modulate a large area of assembly, reminiscent of the catalyst in chemical reactions. This work provides an efficient and flexible approach for the catalysis and precise modulation of molecular assembly. |
| 211. | | Xiaoyu Qi, Luis Alberto Pérez, Jose Mendoza-Carreño, Miquel Garriga, Maria Isabel Alonso, Agustín Mihi Chiral plasmonic superlattices from template-assisted assembly of achiral nanoparticles In: Nature Communications, vol. 16, no. 1687, 2025, (The development of scalable chiral photonic structures is a challenging task. Here, the authors use soft lithography with colloidal plasmonic inks to create chiral plasmonic arrays that enable nanophotonic films with strong chiroptical response.). @article{nokey,
title = {Chiral plasmonic superlattices from template-assisted assembly of achiral nanoparticles},
author = {Xiaoyu Qi and Luis Alberto Pérez and Jose Mendoza-Carreño and Miquel Garriga and Maria Isabel Alonso and Agustín Mihi },
url = {https://www.nature.com/articles/s41467-025-56999-0.pdf},
doi = {10.1038/s41467-025-56999-0},
year = {2025},
date = {2025-02-16},
journal = {Nature Communications},
volume = {16},
number = {1687},
abstract = {The creation of chiral plasmonic architectures combining templates with achiral plasmonic particles leads to strong chiroptical responses that can be finely tuned via the characteristics of the colloidal building blocks. Here we show how elastomeric molds, pre-patterned with a hexagonal array of triskelia motifs, can guide the assembly of ordinary noble metal colloids into chiral plasmonic architectures with strong dichroism values. Under normal incidence, the chiral arrays made with gold and silver colloids showed g-factors of 0.18 and 0.4, respectively. In all cases, increasing the size of the colloid allows tuning the optical properties of the structure in the VIS-NIR range. When a superstrate layer is deposited onto the structures, the extrinsic chirality response of the 2D superlattice is revealed and strongly amplified by the chiral motifs under oblique inspection, leading to g-factors of ± 1.2 at ± 14°. Finally, these chiral plasmonic resonances sustained by the triskelion array are used to produce circularly polarized photoluminescence from achiral organic dyes placed on top with up to 20% of dissymmetry.},
note = {The development of scalable chiral photonic structures is a challenging task. Here, the authors use soft lithography with colloidal plasmonic inks to create chiral plasmonic arrays that enable nanophotonic films with strong chiroptical response.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The creation of chiral plasmonic architectures combining templates with achiral plasmonic particles leads to strong chiroptical responses that can be finely tuned via the characteristics of the colloidal building blocks. Here we show how elastomeric molds, pre-patterned with a hexagonal array of triskelia motifs, can guide the assembly of ordinary noble metal colloids into chiral plasmonic architectures with strong dichroism values. Under normal incidence, the chiral arrays made with gold and silver colloids showed g-factors of 0.18 and 0.4, respectively. In all cases, increasing the size of the colloid allows tuning the optical properties of the structure in the VIS-NIR range. When a superstrate layer is deposited onto the structures, the extrinsic chirality response of the 2D superlattice is revealed and strongly amplified by the chiral motifs under oblique inspection, leading to g-factors of ± 1.2 at ± 14°. Finally, these chiral plasmonic resonances sustained by the triskelion array are used to produce circularly polarized photoluminescence from achiral organic dyes placed on top with up to 20% of dissymmetry. |
| 210. | | Nicolò Canestrari, Diana Nelli, Riccardo Ferrando General theory for packing icosahedral shells into multi-component aggregates In: Nature Communications, vol. 16, no. 1655, 2025, (The icosahedron is the most symmetrical solid structure and is found in metal clusters, colloidal aggregates, viruses and organelles. Here, the authors propose a general theory for the design of multi-component icosahedral aggregates by packing shells of different types.). @article{nokey,
title = {General theory for packing icosahedral shells into multi-component aggregates},
author = {Nicolò Canestrari and Diana Nelli and Riccardo Ferrando },
url = {https://www.nature.com/articles/s41467-025-56952-1.pdf},
doi = {10.1038/s41467-025-56952-1},
year = {2025},
date = {2025-02-15},
journal = {Nature Communications},
volume = {16},
number = {1655},
abstract = {Multi-component aggregates are being intensively researched in various fields because of their highly tunable properties and wide applications. Due to the complex configurational space of these systems, research would greatly benefit from a general theoretical framework for the prediction of stable structures, which, however, is largely incomplete at present. Here we propose a general theory for the construction of multi-component icosahedral structures by assembling concentric shells of different chiral and achiral types, consisting of particles of different sizes. By mapping shell sequences into paths in the hexagonal lattice, we establish simple and general rules for designing a wide variety of magic icosahedral structures, and we evaluate the optimal size-mismatch between particles in the different shells. The predictions of our design strategy are confirmed by molecular dynamics simulations and density functional theory calculations for several multi-component atomic clusters and nanoparticles.},
note = {The icosahedron is the most symmetrical solid structure and is found in metal clusters, colloidal aggregates, viruses and organelles. Here, the authors propose a general theory for the design of multi-component icosahedral aggregates by packing shells of different types.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Multi-component aggregates are being intensively researched in various fields because of their highly tunable properties and wide applications. Due to the complex configurational space of these systems, research would greatly benefit from a general theoretical framework for the prediction of stable structures, which, however, is largely incomplete at present. Here we propose a general theory for the construction of multi-component icosahedral structures by assembling concentric shells of different chiral and achiral types, consisting of particles of different sizes. By mapping shell sequences into paths in the hexagonal lattice, we establish simple and general rules for designing a wide variety of magic icosahedral structures, and we evaluate the optimal size-mismatch between particles in the different shells. The predictions of our design strategy are confirmed by molecular dynamics simulations and density functional theory calculations for several multi-component atomic clusters and nanoparticles. |
| 209. | | Jiapeng Zheng, Yuang Fu, Jing Wang, Wei Zhang, Xinhui Lu, Hai-Qing Lin, Lei Shao, Jianfang Wang Circularly polarized OLEDs from chiral plasmonic nanoparticle-molecule hybrids In: Nature Communications, vol. 16, no. 1658, 2025, (OLEDs that emit circularly polarized light are essential for many technologies. Here, the authors develop OLEDs based on chiral nanoparticles that have large emission dissymmetry factors and high external quantum efficiencies.). @article{nokey,
title = {Circularly polarized OLEDs from chiral plasmonic nanoparticle-molecule hybrids},
author = {Jiapeng Zheng and Yuang Fu and Jing Wang and Wei Zhang and Xinhui Lu and Hai-Qing Lin and Lei Shao and Jianfang Wang },
url = {https://www.nature.com/articles/s41467-025-57000-8.pdf},
doi = {10.1038/s41467-025-57000-8},
year = {2025},
date = {2025-02-15},
journal = {Nature Communications},
volume = {16},
number = {1658},
abstract = {Organic light-emitting diodes (OLEDs) supporting the direct emission of circularly polarized (CP) light are essential for numerous technologies. The realization of CP-OLEDs with large dissymmetry (gEL) factors and high external quantum efficiencies (EQEs) has been accepted as a considerable challenge. Here we demonstrate the realization of efficient CP-OLEDs based on the assembly of chiral plasmonic nanoparticles (NPs) and supramolecular aggregates. The chiral plasmonic NPs serve as the chiral scaffold and chiral optical nanoantenna to modulate the circularly polarized absorption and emission of the supramolecular chromophores. We employ different chiral plasmonic NPs to construct various CP-OLEDs with the emission dominated by chiral excitons or chiral plasmons. The CP-OLED showing a high EQE of 2.5% and a large gEL factor of 0.31 is achieved, as a result of multiscale chirality transfer, plasmonic enhancement, and the suppression of the overshoot effect. The proposed schemes are compatible with the current manufacturing technology of OLEDs. This work demonstrates that chiral plasmonic NPs can be promising candidates in chiral photoelectric devices.},
note = {OLEDs that emit circularly polarized light are essential for many technologies. Here, the authors develop OLEDs based on chiral nanoparticles that have large emission dissymmetry factors and high external quantum efficiencies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Organic light-emitting diodes (OLEDs) supporting the direct emission of circularly polarized (CP) light are essential for numerous technologies. The realization of CP-OLEDs with large dissymmetry (gEL) factors and high external quantum efficiencies (EQEs) has been accepted as a considerable challenge. Here we demonstrate the realization of efficient CP-OLEDs based on the assembly of chiral plasmonic nanoparticles (NPs) and supramolecular aggregates. The chiral plasmonic NPs serve as the chiral scaffold and chiral optical nanoantenna to modulate the circularly polarized absorption and emission of the supramolecular chromophores. We employ different chiral plasmonic NPs to construct various CP-OLEDs with the emission dominated by chiral excitons or chiral plasmons. The CP-OLED showing a high EQE of 2.5% and a large gEL factor of 0.31 is achieved, as a result of multiscale chirality transfer, plasmonic enhancement, and the suppression of the overshoot effect. The proposed schemes are compatible with the current manufacturing technology of OLEDs. This work demonstrates that chiral plasmonic NPs can be promising candidates in chiral photoelectric devices. |
| 208. | | Zhen Sun, Yao Zhang, Zezhou Li, Zhiheng Xie, Yiheng Dai, Xuanxuan Du, Colin Ophus, Jihan Zhou Strain release by 3D atomic misfit in fivefold twinned icosahedral nanoparticles with amorphization and dislocations In: Nature Communications, vol. 16, no. 1595, 2025. @article{nokey,
title = {Strain release by 3D atomic misfit in fivefold twinned icosahedral nanoparticles with amorphization and dislocations},
author = {Zhen Sun and Yao Zhang and Zezhou Li and Zhiheng Xie and Yiheng Dai and Xuanxuan Du and Colin Ophus and Jihan Zhou },
url = {https://www.nature.com/articles/s41467-025-56842-6.pdf},
doi = {10.1038/s41467-025-56842-6},
year = {2025},
date = {2025-02-13},
journal = {Nature Communications},
volume = {16},
number = {1595},
abstract = {Multiple twinning to form fivefold twinned nanoparticles in crystal growth is common and has attracted broad attention ranging from crystallography research to physical chemistry and materials science. Lattice-misfit strain and defects in multiple twinned nanoparticles (MTP) are key to understand and tailor their electronic properties. However, the structural defects and related strain distributions in MTPs are poorly understood in three dimensions (3D). Here, we show the 3D atomic misfit and strain relief mechanism in fivefold twinned icosahedral nanoparticles with amorphization and dislocations by using atomic resolution electron tomography. We discover a two-sided heterogeneity in variety of structural characteristics. A nearly ideal crystallographic fivefold face is always found opposite to a less ordered face, forming Janus-like icosahedral nanoparticles with two distinct hemispheres. The disordered amorphous domains release a large amount of strain. Molecular dynamics simulations further reveal the Janus-like icosahedral nanoparticles are prevalent in the MTPs formed in liquid-solid phase transition. This work provides insights on the atomistic models for the modelling of formation mechanisms of fivefold twinned structures and computational simulations of lattice distortions and defects. We anticipate it will inspire future studies on fundamental problems such as twin boundary migration and kinetics of structures in 3D at atomic level.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Multiple twinning to form fivefold twinned nanoparticles in crystal growth is common and has attracted broad attention ranging from crystallography research to physical chemistry and materials science. Lattice-misfit strain and defects in multiple twinned nanoparticles (MTP) are key to understand and tailor their electronic properties. However, the structural defects and related strain distributions in MTPs are poorly understood in three dimensions (3D). Here, we show the 3D atomic misfit and strain relief mechanism in fivefold twinned icosahedral nanoparticles with amorphization and dislocations by using atomic resolution electron tomography. We discover a two-sided heterogeneity in variety of structural characteristics. A nearly ideal crystallographic fivefold face is always found opposite to a less ordered face, forming Janus-like icosahedral nanoparticles with two distinct hemispheres. The disordered amorphous domains release a large amount of strain. Molecular dynamics simulations further reveal the Janus-like icosahedral nanoparticles are prevalent in the MTPs formed in liquid-solid phase transition. This work provides insights on the atomistic models for the modelling of formation mechanisms of fivefold twinned structures and computational simulations of lattice distortions and defects. We anticipate it will inspire future studies on fundamental problems such as twin boundary migration and kinetics of structures in 3D at atomic level. |
| 207. | | Cui-Mi Shi, Haolin Lu, Jin-Yun Wang, Guankui Long, Liang-Jin Xu, Zhong-Ning Chen Stepwise amplification of circularly polarized luminescence in indium-based metal halides by regulating their structural dimension In: Nature Communications, vol. 16, no. 1505, 2025, (Designing efficient circularly polarized light sources requires a balance between the photoluminescence quantum yield and the luminescence dissymmetry factor. Here, the authors develop indium-based chiral metal halides with efficient CPL characteristics by modulating their structural dimensions.). @article{nokey,
title = {Stepwise amplification of circularly polarized luminescence in indium-based metal halides by regulating their structural dimension},
author = {Cui-Mi Shi and Haolin Lu and Jin-Yun Wang and Guankui Long and Liang-Jin Xu and Zhong-Ning Chen },
url = {https://www.nature.com/articles/s41467-025-56394-9.pdf},
doi = {10.1038/s41467-025-56394-9},
year = {2025},
date = {2025-02-10},
journal = {Nature Communications},
volume = {16},
number = {1505},
abstract = {The pursuit of chiral lead-free metal halides with both high photoluminescence quantum yield (PLQY) and large luminescence dissymmetry factor (glum) remains a priority for designing efficient circularly polarized light sources. However, a tradeoff exists between PLQY and glum in chiral materials due to the mismatched electric (μ) and magnetic transition dipole moment (m). Herein, we address this contradiction and develop the efficient circularly polarized luminescence (CPL) emitters through structural dimension modulation. By tuning the size and polarization of chiral organic cations and employing the cascade cationic insertion strategy, 0D, 1D and 3D indium-based chiral metal halides are constructed. These hybrids exhibit self-trapped excitons emission with near-unity PLQY, while the |glum| boosts exponentially from 10−3 to nearly 10−1 as the structural dimension increases from 0D to 3D, and the highest |glum| of 0.89 × 10−1 has been achieved. Structural analysis and theoretical calculation indicate the increased structural dimension promotes the formation of helical structure and enlarges magnetic transition dipole moment, thus resulting in improved CPL performance. Our research provides valuable insights on the relationship between glum and structural dimension, thus will advance the development of efficient CPL-active materials for practical applications.},
note = {Designing efficient circularly polarized light sources requires a balance between the photoluminescence quantum yield and the luminescence dissymmetry factor. Here, the authors develop indium-based chiral metal halides with efficient CPL characteristics by modulating their structural dimensions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The pursuit of chiral lead-free metal halides with both high photoluminescence quantum yield (PLQY) and large luminescence dissymmetry factor (glum) remains a priority for designing efficient circularly polarized light sources. However, a tradeoff exists between PLQY and glum in chiral materials due to the mismatched electric (μ) and magnetic transition dipole moment (m). Herein, we address this contradiction and develop the efficient circularly polarized luminescence (CPL) emitters through structural dimension modulation. By tuning the size and polarization of chiral organic cations and employing the cascade cationic insertion strategy, 0D, 1D and 3D indium-based chiral metal halides are constructed. These hybrids exhibit self-trapped excitons emission with near-unity PLQY, while the |glum| boosts exponentially from 10−3 to nearly 10−1 as the structural dimension increases from 0D to 3D, and the highest |glum| of 0.89 × 10−1 has been achieved. Structural analysis and theoretical calculation indicate the increased structural dimension promotes the formation of helical structure and enlarges magnetic transition dipole moment, thus resulting in improved CPL performance. Our research provides valuable insights on the relationship between glum and structural dimension, thus will advance the development of efficient CPL-active materials for practical applications. |
| 206. | | Tianyi Wu, Sina Kheiri, Riley J. Hickman, Huachen Tao, Tony C. Wu, Zhi-Bo Yang, Xin Ge, Wei Zhang, Milad Abolhasani, Kun Liu, Alan Aspuru-Guzik, Eugenia Kumacheva Self-driving lab for the photochemical synthesis of plasmonic nanoparticles with targeted structural and optical properties In: Nature Communications, vol. 16, no. 1473, 2025, (The automated synthesis of plasmonic nanoparticles with on-demand properties is a challenging task. Here the authors integrate a fluidic reactor, real-time characterization, and machine learning in a self-driven lab for the photochemical synthesis of nanoparticles with targeted properties.). @article{nokey,
title = {Self-driving lab for the photochemical synthesis of plasmonic nanoparticles with targeted structural and optical properties},
author = {Tianyi Wu and Sina Kheiri and Riley J. Hickman and Huachen Tao and Tony C. Wu and Zhi-Bo Yang and Xin Ge and Wei Zhang and Milad Abolhasani and Kun Liu and Alan Aspuru-Guzik and Eugenia Kumacheva },
url = {https://www.nature.com/articles/s41467-025-56788-9.pdf},
doi = {10.1038/s41467-025-56788-9},
year = {2025},
date = {2025-02-08},
journal = {Nature Communications},
volume = {16},
number = {1473},
abstract = {Many applications of plasmonic nanoparticles require precise control of their optical properties that are governed by nanoparticle dimensions, shape, morphology and composition. Finding reaction conditions for the synthesis of nanoparticles with targeted characteristics is a time-consuming and resource-intensive trial-and-error process, however closed-loop nanoparticle synthesis enables the accelerated exploration of large chemical spaces without human intervention. Here, we introduce the Autonomous Fluidic Identification and Optimization Nanochemistry (AFION) self-driving lab that integrates a microfluidic reactor, in-flow spectroscopic nanoparticle characterization, and machine learning for the exploration and optimization of the multidimensional chemical space for the photochemical synthesis of plasmonic nanoparticles. By targeting spectroscopic nanoparticle properties, the AFION lab identifies reaction conditions for the synthesis of different types of nanoparticles with designated shapes, morphologies, and compositions. Data analysis provides insight into the role of reaction conditions for the synthesis of the targeted nanoparticle type. This work shows that the AFION lab is an effective exploration platform for on-demand synthesis of plasmonic nanoparticles.},
note = {The automated synthesis of plasmonic nanoparticles with on-demand properties is a challenging task. Here the authors integrate a fluidic reactor, real-time characterization, and machine learning in a self-driven lab for the photochemical synthesis of nanoparticles with targeted properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Many applications of plasmonic nanoparticles require precise control of their optical properties that are governed by nanoparticle dimensions, shape, morphology and composition. Finding reaction conditions for the synthesis of nanoparticles with targeted characteristics is a time-consuming and resource-intensive trial-and-error process, however closed-loop nanoparticle synthesis enables the accelerated exploration of large chemical spaces without human intervention. Here, we introduce the Autonomous Fluidic Identification and Optimization Nanochemistry (AFION) self-driving lab that integrates a microfluidic reactor, in-flow spectroscopic nanoparticle characterization, and machine learning for the exploration and optimization of the multidimensional chemical space for the photochemical synthesis of plasmonic nanoparticles. By targeting spectroscopic nanoparticle properties, the AFION lab identifies reaction conditions for the synthesis of different types of nanoparticles with designated shapes, morphologies, and compositions. Data analysis provides insight into the role of reaction conditions for the synthesis of the targeted nanoparticle type. This work shows that the AFION lab is an effective exploration platform for on-demand synthesis of plasmonic nanoparticles. |
| 205. | | Makusu Tsutsui, Wei-Lun Hsu, Chien Hsu, Denis Garoli, Shukun Weng, Hirofumi Daiguji, Tomoji Kawai Transmembrane voltage-gated nanopores controlled by electrically tunable in-pore chemistry In: Nature Communications, vol. 16, no. 1089, 2025, (Ion channels in cell membranes open and close in response to electrical stimuli, playing a critical role in cellular signaling and homeostasis. Here, the authors show a similar gating functionality in solid-state nanopores, achieved through transmembrane voltage control of in-pore chemistry.). @article{nokey,
title = {Transmembrane voltage-gated nanopores controlled by electrically tunable in-pore chemistry},
author = {Makusu Tsutsui and Wei-Lun Hsu and Chien Hsu and Denis Garoli and Shukun Weng and Hirofumi Daiguji and Tomoji Kawai },
url = {https://www.nature.com/articles/s41467-025-56052-0.pdf},
doi = {10.1038/s41467-025-56052-0},
year = {2025},
date = {2025-02-05},
journal = {Nature Communications},
volume = {16},
number = {1089},
abstract = {Gating is a fundamental process in ion channels configured to open and close in response to specific stimuli such as voltage across cell membranes thereby enabling the excitability of neurons. Here we report on voltage-gated solid-state nanopores by electrically tunable chemical reactions. We demonstrate repetitive precipitation and dissolution of metal phosphates in a pore through manipulations of cation flow by transmembrane voltage. Under negative voltages, precipitates grow to reduce ionic current by occluding the nanopore, while inverting the voltage polarity dissolves the phosphate compounds reopening the pore to ionic flux. Reversible actuation of these physicochemical processes creates a nanofluidic diode of rectification ratio exceeding 40000. The dynamic nature of the in-pore reactions also facilitates a memristor of sub-nanowatt power consumption. Leveraging chemical degrees of freedom, the present method may be useful for creating iontronic circuits of tunable characteristics toward neuromorphic systems.},
note = {Ion channels in cell membranes open and close in response to electrical stimuli, playing a critical role in cellular signaling and homeostasis. Here, the authors show a similar gating functionality in solid-state nanopores, achieved through transmembrane voltage control of in-pore chemistry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gating is a fundamental process in ion channels configured to open and close in response to specific stimuli such as voltage across cell membranes thereby enabling the excitability of neurons. Here we report on voltage-gated solid-state nanopores by electrically tunable chemical reactions. We demonstrate repetitive precipitation and dissolution of metal phosphates in a pore through manipulations of cation flow by transmembrane voltage. Under negative voltages, precipitates grow to reduce ionic current by occluding the nanopore, while inverting the voltage polarity dissolves the phosphate compounds reopening the pore to ionic flux. Reversible actuation of these physicochemical processes creates a nanofluidic diode of rectification ratio exceeding 40000. The dynamic nature of the in-pore reactions also facilitates a memristor of sub-nanowatt power consumption. Leveraging chemical degrees of freedom, the present method may be useful for creating iontronic circuits of tunable characteristics toward neuromorphic systems. |
| 204. | | Akira Takakura, Taishi Nishihara, Koji Harano, Ovidiu Cretu, Takeshi Tanaka, Hiromichi Kataura, Yuhei Miyauchi Coalescence of carbon nanotubes while preserving the chiral angles In: Nature Communications, vol. 16, no. 1093, 2025, (Coalescence of macromolecules with structural precision is a challenging reaction. Here, the authors demonstrate the chiral-angle conserved coalescence of carbon nanotubes with a yield of up to 20%-40%, which enables the selective synthesis of large-diameter nanotubes.). @article{nokey,
title = {Coalescence of carbon nanotubes while preserving the chiral angles},
author = {Akira Takakura and Taishi Nishihara and Koji Harano and Ovidiu Cretu and Takeshi Tanaka and Hiromichi Kataura and Yuhei Miyauchi },
url = {https://www.nature.com/articles/s41467-025-56389-6.pdf},
doi = {10.1038/s41467-025-56389-6},
year = {2025},
date = {2025-02-05},
urldate = {2025-02-05},
journal = {Nature Communications},
volume = {16},
number = {1093},
abstract = {Atomically precise coalescence of graphitic nanocarbon molecules is one of the most challenging reactions in sp2 carbon chemistry. Here, we demonstrate that two carbon nanotubes with the same chiral indices (n, m) are efficiently coalesced into a single (2n, 2 m) nanotube with preserved chiral angles via heat treatment at less than 1000 °C. The (2n, 2 m) nanotubes constitute up to ≈ 20%–40% of the final sample in the most efficient case. Additional optical absorption peaks of the (2n, 2 m) nanotubes emerge, indicating that the reaction occurs over the entire sample. The reaction efficiency strongly depends on the chiral angle, implying that C–C bond cleavage and recombination occurs sequentially. Furthermore, the reaction occurs efficiently even at 600 °C in an atmosphere containing trace amounts of oxygen. These findings offer routes for the structure-selective synthesis of large-diameter nanotubes and modification of the properties of nanotube assemblies via postprocessing.},
note = {Coalescence of macromolecules with structural precision is a challenging reaction. Here, the authors demonstrate the chiral-angle conserved coalescence of carbon nanotubes with a yield of up to 20%-40%, which enables the selective synthesis of large-diameter nanotubes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Atomically precise coalescence of graphitic nanocarbon molecules is one of the most challenging reactions in sp2 carbon chemistry. Here, we demonstrate that two carbon nanotubes with the same chiral indices (n, m) are efficiently coalesced into a single (2n, 2 m) nanotube with preserved chiral angles via heat treatment at less than 1000 °C. The (2n, 2 m) nanotubes constitute up to ≈ 20%–40% of the final sample in the most efficient case. Additional optical absorption peaks of the (2n, 2 m) nanotubes emerge, indicating that the reaction occurs over the entire sample. The reaction efficiency strongly depends on the chiral angle, implying that C–C bond cleavage and recombination occurs sequentially. Furthermore, the reaction occurs efficiently even at 600 °C in an atmosphere containing trace amounts of oxygen. These findings offer routes for the structure-selective synthesis of large-diameter nanotubes and modification of the properties of nanotube assemblies via postprocessing. |
| 203. | | Qiqi Yang, Antonio Virgilio Failla, Petri Turunen, Ana Mateos-Maroto, Meiyu Gai, Werner Zuschratter, Sophia Westendorf, Márton Gelléri, Qiang Chen, Goudappagouda, Hao Zhao, Xingfu Zhu, Svenja Morsbach, Marcus Scheele, Wei Yan, Katharina Landfester, Ryota Kabe, Mischa Bonn, Akimitsu Narita, Xiaomin Liu Reactivatable stimulated emission depletion microscopy using fluorescence-recoverable nanographene In: Nature Communications, vol. 16, no. 1341, 2025, (Stimulated emission depletion (STED) microscopy is compromised by the trade-off between resolution and photobleaching. Here, the authors present ReSTED, a reactivatable STED microscopy using fluorescence-recoverable nanographene that enables hour-long, super-resolution 3D imaging without bleaching.). @article{nokey,
title = {Reactivatable stimulated emission depletion microscopy using fluorescence-recoverable nanographene},
author = {Qiqi Yang and Antonio Virgilio Failla and Petri Turunen and Ana Mateos-Maroto and Meiyu Gai and Werner Zuschratter and Sophia Westendorf and Márton Gelléri and Qiang Chen and Goudappagouda and Hao Zhao and Xingfu Zhu and Svenja Morsbach and Marcus Scheele and Wei Yan and Katharina Landfester and Ryota Kabe and Mischa Bonn and Akimitsu Narita and Xiaomin Liu },
doi = {10.1038/s41467-025-56401-z},
year = {2025},
date = {2025-02-04},
urldate = {2025-02-04},
journal = {Nature Communications},
volume = {16},
number = {1341},
abstract = {Stimulated emission depletion (STED) microscopy, a key optical super-resolution imaging method, has extended our ability to view details to resolution levels of tens of nanometers. Its resolution depends on fluorophore de-excitation efficiency, and increases with depletion laser power. However, high-power irradiation permanently turns off the fluorescence due to photo-bleaching of the fluorophores. As a result, there is a trade-off between spatial resolution and imaging time. Here, we overcome this limitation by introducing reactivatable STED (ReSTED) based on the photophysical properties of the nanographene dibenzo[hi,st]ovalene (DBOV). In contrast to the photo-induced decomposition of other fluorophores, the fluorescence of DBOV is only temporarily deactivated and can be reactivated by near-infrared light (including the 775 nm depletion beam). As a result, this fluorophore allows for hours-long, high-resolution 3D STED imaging, greatly expanding the applications of STED microscopy.},
note = {Stimulated emission depletion (STED) microscopy is compromised by the trade-off between resolution and photobleaching. Here, the authors present ReSTED, a reactivatable STED microscopy using fluorescence-recoverable nanographene that enables hour-long, super-resolution 3D imaging without bleaching.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Stimulated emission depletion (STED) microscopy, a key optical super-resolution imaging method, has extended our ability to view details to resolution levels of tens of nanometers. Its resolution depends on fluorophore de-excitation efficiency, and increases with depletion laser power. However, high-power irradiation permanently turns off the fluorescence due to photo-bleaching of the fluorophores. As a result, there is a trade-off between spatial resolution and imaging time. Here, we overcome this limitation by introducing reactivatable STED (ReSTED) based on the photophysical properties of the nanographene dibenzo[hi,st]ovalene (DBOV). In contrast to the photo-induced decomposition of other fluorophores, the fluorescence of DBOV is only temporarily deactivated and can be reactivated by near-infrared light (including the 775 nm depletion beam). As a result, this fluorophore allows for hours-long, high-resolution 3D STED imaging, greatly expanding the applications of STED microscopy. |
| 202. | | Vivek Yadav, Arijit Jana, Swetashree Acharya, Sami Malola, Harshita Nagar, Ankit Sharma, Amoghavarsha Ramachandra Kini, Sudhadevi Antharjanam, Jan Machacek, Kumaran Nair Valsala Devi Adarsh, Tomas Base, Hannu Häkkinen, Thalappil Pradeep Site-specific substitution in atomically precise carboranethiol-protected nanoclusters and concomitant changes in electronic properties In: Nature Communications, vol. 16, no. 1197, 2025, (Tuning the structure and composition of atomically precise metal nanoclusters leads to property changes which, however, are still poorly understood. Here, the authors synthesize precisely substituted analogues of Ag17 clusters and study the changes in luminescence and electronic properties.). @article{nokey,
title = {Site-specific substitution in atomically precise carboranethiol-protected nanoclusters and concomitant changes in electronic properties},
author = {Vivek Yadav and Arijit Jana and Swetashree Acharya and Sami Malola and Harshita Nagar and Ankit Sharma and Amoghavarsha Ramachandra Kini and Sudhadevi Antharjanam and Jan Machacek and Kumaran Nair Valsala Devi Adarsh and Tomas Base and Hannu Häkkinen and Thalappil Pradeep },
url = {https://www.nature.com/articles/s41467-025-56385-w.pdf},
doi = {10.1038/s41467-025-56385-w},
year = {2025},
date = {2025-01-30},
journal = {Nature Communications},
volume = {16},
number = {1197},
abstract = {We report the synthesis of [Ag17(o1-CBT)12]3- abbreviated as Ag17, a stable 8e⁻ anionic cluster with a unique Ag@Ag12@Ag4 core-shell structure, where o1-CBT is ortho-carborane-1-thiol. By substituting Ag atoms with Au and/or Cu at specific sites we created isostructural clusters [AuAg16(o1-CBT)12]3- (AuAg16), [Ag13Cu4(o1-CBT)12]3- (Ag13Cu4) and [AuAg12Cu4(o1-CBT)12]3- (AuAg12Cu4). These substitutions make systematic modulation of their structural and electronic properties. We show that Au preferentially occupies the core, while Cu localizes in the tetrahedral shell, influencing stability and structural diversity of the clusters. The band gap expands systematically (2.09 eV for Ag17 to 2.28 eV for AuAg12Cu4), altering optical absorption and emission. Ultrafast optical measurements reveal longer excited-state lifetimes for Cu-containing clusters, highlighting the effect of heteroatom incorporation. These results demonstrate a tunable platform for designing nanoclusters with tailored electronic properties, with implications for optoelectronics and catalysis.},
note = {Tuning the structure and composition of atomically precise metal nanoclusters leads to property changes which, however, are still poorly understood. Here, the authors synthesize precisely substituted analogues of Ag17 clusters and study the changes in luminescence and electronic properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We report the synthesis of [Ag17(o1-CBT)12]3- abbreviated as Ag17, a stable 8e⁻ anionic cluster with a unique Ag@Ag12@Ag4 core-shell structure, where o1-CBT is ortho-carborane-1-thiol. By substituting Ag atoms with Au and/or Cu at specific sites we created isostructural clusters [AuAg16(o1-CBT)12]3- (AuAg16), [Ag13Cu4(o1-CBT)12]3- (Ag13Cu4) and [AuAg12Cu4(o1-CBT)12]3- (AuAg12Cu4). These substitutions make systematic modulation of their structural and electronic properties. We show that Au preferentially occupies the core, while Cu localizes in the tetrahedral shell, influencing stability and structural diversity of the clusters. The band gap expands systematically (2.09 eV for Ag17 to 2.28 eV for AuAg12Cu4), altering optical absorption and emission. Ultrafast optical measurements reveal longer excited-state lifetimes for Cu-containing clusters, highlighting the effect of heteroatom incorporation. These results demonstrate a tunable platform for designing nanoclusters with tailored electronic properties, with implications for optoelectronics and catalysis. |
| 201. | | Brian Nosek, Christine Mummery, Leonardo Scarabelli, Vitaly Podzorov Reproducibility and transparency: what’s going on and how can we help (Q&A article) In: Nature Communications, vol. 16, no. 1082, 2025, (Problems with experimental reproducibility affect every field of science. However, the opinions on the causes of the reproducibility “crisis” and how we all can help vary amongst fields as well as individual scientists. Here, we talk to experts from different fields of science to get their insights on this endemic issue.). @article{nokey,
title = {Reproducibility and transparency: what’s going on and how can we help (Q&A article)},
author = {Brian Nosek and Christine Mummery and Leonardo Scarabelli and Vitaly Podzorov },
url = {https://www.nature.com/articles/s41467-024-54614-2.pdf},
doi = {10.1038/s41467-024-54614-2},
year = {2025},
date = {2025-01-29},
urldate = {2024-12-04},
journal = {Nature Communications},
volume = {16},
number = {1082},
abstract = {Problems with experimental reproducibility affect every field of science. However, the opinions on the causes of the reproducibility “crisis” and how we all can help vary amongst fields as well as individual scientists. Here, we talk to experts from different fields of science to get their insights on this endemic issue. Professor Brian Nosek is a social psychologist at the University of Virginia and executive director of the Center for Open Science. Professor Christine Mummery is a developmental biologist at Leiden University Medical Center and the former President of the International Society of Stem Cell Research. Dr Leonardo Scarabelli is a chemist and group leader at the University of Cantabria. Professor Vitaly Podzorov is a physicist at Rutgers, the State University of New Jersey, and current Donald H. Jacobs Chair in Applied Physics.},
note = {Problems with experimental reproducibility affect every field of science. However, the opinions on the causes of the reproducibility “crisis” and how we all can help vary amongst fields as well as individual scientists. Here, we talk to experts from different fields of science to get their insights on this endemic issue.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Problems with experimental reproducibility affect every field of science. However, the opinions on the causes of the reproducibility “crisis” and how we all can help vary amongst fields as well as individual scientists. Here, we talk to experts from different fields of science to get their insights on this endemic issue. Professor Brian Nosek is a social psychologist at the University of Virginia and executive director of the Center for Open Science. Professor Christine Mummery is a developmental biologist at Leiden University Medical Center and the former President of the International Society of Stem Cell Research. Dr Leonardo Scarabelli is a chemist and group leader at the University of Cantabria. Professor Vitaly Podzorov is a physicist at Rutgers, the State University of New Jersey, and current Donald H. Jacobs Chair in Applied Physics. |
| 200. | | Sungsu Kang, Joodeok Kim, Sungin Kim, Hoje Chun, Junyoung Heo, Cyril F. Reboul, Rubén Meana-Pañeda, Cong T. S. Van, Hyesung Choi, Yunseo Lee, Jinho Rhee, Minyoung Lee, Dohun Kang, Byung Hyo Kim, Taeghwan Hyeon, Byungchan Han, Peter Ercius, Won Chul Lee, Hans Elmlund, Jungwon Park Time-resolved Brownian tomography of single nanocrystals in liquid during oxidative etching In: Nature Communications, vol. 16, no. 1158, 2025, (Atomic structural changes in nanocrystals are still poorly understood. Here, the authors introduce time-resolved Brownian tomography to directly detect 3D atomic changes in Pt nanocrystals during oxidative etching and reveal disorder at sizes of ≈1 nm.). @article{nokey,
title = {Time-resolved Brownian tomography of single nanocrystals in liquid during oxidative etching},
author = {Sungsu Kang and Joodeok Kim and Sungin Kim and Hoje Chun and Junyoung Heo and Cyril F. Reboul and Rubén Meana-Pañeda and Cong T. S. Van and Hyesung Choi and Yunseo Lee and Jinho Rhee and Minyoung Lee and Dohun Kang and Byung Hyo Kim and Taeghwan Hyeon and Byungchan Han and Peter Ercius and Won Chul Lee and Hans Elmlund and Jungwon Park },
url = {https://www.nature.com/articles/s41467-025-56476-8.pdf},
doi = {10.1038/s41467-025-56476-8},
year = {2025},
date = {2025-01-29},
journal = {Nature Communications},
volume = {16},
number = {1158},
abstract = {Colloidal nanocrystals inherently undergo structural changes during chemical reactions. The robust structure-property relationships, originating from their nanoscale dimensions, underscore the significance of comprehending the dynamic structural behavior of nanocrystals in reactive chemical media. Moreover, the complexity and heterogeneity inherent in their atomic structures require tracking of structural transitions in individual nanocrystals at three-dimensional (3D) atomic resolution. In this study, we introduce the method of time-resolved Brownian tomography to investigate the temporal evolution of the 3D atomic structures of individual nanocrystals in solution. The methodology is applied to examine the atomic-level structural transformations of Pt nanocrystals during oxidative etching. The time-resolved 3D atomic maps reveal the structural evolution of dissolving Pt nanocrystals, transitioning from a crystalline to a disordered structure. Our study demonstrates the emergence of a phase at the nanometer length scale that has received less attention in bulk thermodynamics.},
note = {Atomic structural changes in nanocrystals are still poorly understood. Here, the authors introduce time-resolved Brownian tomography to directly detect 3D atomic changes in Pt nanocrystals during oxidative etching and reveal disorder at sizes of ≈1 nm.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Colloidal nanocrystals inherently undergo structural changes during chemical reactions. The robust structure-property relationships, originating from their nanoscale dimensions, underscore the significance of comprehending the dynamic structural behavior of nanocrystals in reactive chemical media. Moreover, the complexity and heterogeneity inherent in their atomic structures require tracking of structural transitions in individual nanocrystals at three-dimensional (3D) atomic resolution. In this study, we introduce the method of time-resolved Brownian tomography to investigate the temporal evolution of the 3D atomic structures of individual nanocrystals in solution. The methodology is applied to examine the atomic-level structural transformations of Pt nanocrystals during oxidative etching. The time-resolved 3D atomic maps reveal the structural evolution of dissolving Pt nanocrystals, transitioning from a crystalline to a disordered structure. Our study demonstrates the emergence of a phase at the nanometer length scale that has received less attention in bulk thermodynamics. |
| 199. | | Yuanyang Xie, Alexey V. Krasavin, Diane J. Roth, Anatoly V. Zayats Unidirectional chiral scattering from single enantiomeric plasmonic nanoparticles In: Nature Communications, vol. 16, no. 1125, 2025, (Controlling the directional scattering of chiral light at the nanoscale is important for optical technologies. Here, the authors present a concept of rotating chiral dipoles to achieve unidirectional enantio-sensitive chiral scattering.). @article{nokey,
title = {Unidirectional chiral scattering from single enantiomeric plasmonic nanoparticles},
author = {Yuanyang Xie and Alexey V. Krasavin and Diane J. Roth and Anatoly V. Zayats },
url = {https://www.nature.com/articles/s41467-024-55277-9.pdf},
doi = {10.1038/s41467-024-55277-9},
year = {2025},
date = {2025-01-28},
journal = {Nature Communications},
volume = {16},
number = {1125},
abstract = {Controlling scattering and routing of chiral light at the nanoscale is important for optical information processing and imaging, quantum technologies as well as optical manipulation. Here, we introduce a concept of rotating chiral dipoles in order to achieve unidirectional chiral scattering. Implementing this concept by engineering multipole excitations in helicoidal plasmonic nanoparticles, we experimentally demonstrate enantio-sensitive and highly-directional forward scattering of circularly polarised light. The intensity of this highly-directional scattering is defined by the mutual relation between the handedness of the incident light and the chirality of the structure. The concept of rotating chiral dipoles offers numerous opportunities for engineering scattering from chiral nanostructures and optical nano-antennas paving the way for innovative designs and applications of chiral light-matter interactions.},
note = {Controlling the directional scattering of chiral light at the nanoscale is important for optical technologies. Here, the authors present a concept of rotating chiral dipoles to achieve unidirectional enantio-sensitive chiral scattering.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Controlling scattering and routing of chiral light at the nanoscale is important for optical information processing and imaging, quantum technologies as well as optical manipulation. Here, we introduce a concept of rotating chiral dipoles in order to achieve unidirectional chiral scattering. Implementing this concept by engineering multipole excitations in helicoidal plasmonic nanoparticles, we experimentally demonstrate enantio-sensitive and highly-directional forward scattering of circularly polarised light. The intensity of this highly-directional scattering is defined by the mutual relation between the handedness of the incident light and the chirality of the structure. The concept of rotating chiral dipoles offers numerous opportunities for engineering scattering from chiral nanostructures and optical nano-antennas paving the way for innovative designs and applications of chiral light-matter interactions. |
| 198. | | Tong Zhang, Yuanbiao Tong, Chenxinyu Pan, Jun Pei, Xiaomeng Wang, Tao Liu, Binglun Yin, Pan Wang, Yang Gao, Limin Tong, Wei Yang Challenging the ideal strength limit in single-crystalline gold nanoflakes through phase engineering In: Nature Communications, vol. 16, no. 926, 2025, (Gold with FCC phase is very ductile, while the yield strength is well below the theoretical limit. Here, the authors report on the mechanical properties of single-crystalline gold nanoflakes with HCP phase that illustrate the benefits of phase engineering.). @article{nokey,
title = {Challenging the ideal strength limit in single-crystalline gold nanoflakes through phase engineering},
author = {Tong Zhang and Yuanbiao Tong and Chenxinyu Pan and Jun Pei and Xiaomeng Wang and Tao Liu and Binglun Yin and Pan Wang and Yang Gao and Limin Tong and Wei Yang },
url = {https://www.nature.com/articles/s41467-025-56047-x.pdf},
doi = {10.1038/s41467-025-56047-x},
year = {2025},
date = {2025-01-22},
urldate = {2025-01-22},
journal = {Nature Communications},
volume = {16},
number = {926},
abstract = {Materials usually fracture before reaching their ideal strength limits. Meanwhile, materials with high strength generally have poor ductility, and vice versa. For example, gold with the conventional face-centered cubic (FCC) phase is highly ductile while the yield strength (~102 MPa) is significantly lower than its ideal theoretical limit. Here, through phase engineering, we show that defect-free single-crystalline gold nanoflakes with the hexagonal close-packed (HCP) phase can exhibit a strength of 6.0 GPa, which is beyond the ideal theoretical limit of the conventional FCC counterpart. The lattice structure is thickness-dependent and the FCC-HCP phase transformation happens in the range of 11–13 nm. Suspended-nanoindentations based on atomic force microscopy (AFM) show that the Young’s modulus and tensile strength are also thickness-and phase- dependent. The maximum strength is reached in HCP nanoflakes thinner than 10 nm. First-principles and molecular dynamics (MD) calculations demonstrate that the mechanical properties arise from the unconventional HCP structure as well as the strong surface effect. Our study provides valuable insights into the fabrication of nanometals with extraordinary mechanical properties through phase engineering.},
note = {Gold with FCC phase is very ductile, while the yield strength is well below the theoretical limit. Here, the authors report on the mechanical properties of single-crystalline gold nanoflakes with HCP phase that illustrate the benefits of phase engineering.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Materials usually fracture before reaching their ideal strength limits. Meanwhile, materials with high strength generally have poor ductility, and vice versa. For example, gold with the conventional face-centered cubic (FCC) phase is highly ductile while the yield strength (~102 MPa) is significantly lower than its ideal theoretical limit. Here, through phase engineering, we show that defect-free single-crystalline gold nanoflakes with the hexagonal close-packed (HCP) phase can exhibit a strength of 6.0 GPa, which is beyond the ideal theoretical limit of the conventional FCC counterpart. The lattice structure is thickness-dependent and the FCC-HCP phase transformation happens in the range of 11–13 nm. Suspended-nanoindentations based on atomic force microscopy (AFM) show that the Young’s modulus and tensile strength are also thickness-and phase- dependent. The maximum strength is reached in HCP nanoflakes thinner than 10 nm. First-principles and molecular dynamics (MD) calculations demonstrate that the mechanical properties arise from the unconventional HCP structure as well as the strong surface effect. Our study provides valuable insights into the fabrication of nanometals with extraordinary mechanical properties through phase engineering. |
| 197. | | Rafal Zuzak, Pawel Dabczynski, Jesús Castro-Esteban, José Ignacio Martínez, Mads Engelund, Dolores Pérez, Diego Peña, Szymon Godlewski Cyclodehydrogenation of molecular nanographene precursors catalyzed by atomic hydrogen In: Nature Communications, vol. 16, no. 691, 2025, (On-surface synthesis of nanographenes proceeds differently on metals than on semiconductors or insulating substrates. Here, the authors perform substrate type-independent chemistry with atomic hydrogen acting as a catalyst in the intramolecular cyclodehydrogenation reaction of polyarenes, yielding atomically precise nanographenes.). @article{nokey,
title = {Cyclodehydrogenation of molecular nanographene precursors catalyzed by atomic hydrogen},
author = {Rafal Zuzak and Pawel Dabczynski and Jesús Castro-Esteban and José Ignacio Martínez and Mads Engelund and Dolores Pérez and Diego Peña and Szymon Godlewski },
url = {https://www.nature.com/articles/s41467-024-54774-1.pdf},
doi = {10.1038/s41467-024-54774-1},
year = {2025},
date = {2025-01-15},
journal = {Nature Communications},
volume = {16},
number = {691},
abstract = {Atomically precise synthesis of graphene nanostructures on semiconductors and insulators has been a formidable challenge. In particular, the metallic substrates needed to catalyze cyclodehydrogenative planarization reactions limit subsequent applications that exploit the electronic and/or magnetic structure of graphene derivatives. Here, we introduce a protocol in which an on-surface reaction is initiated and carried out regardless of the substrate type. We demonstrate that, counterintuitively, atomic hydrogen can play the role of a catalyst in the cyclodehydrogenative planarization reaction. The high efficiency of the method is demonstrated by the nanographene synthesis on metallic Au, semiconducting TiO2, Ge:H, as well as on inert and insulating Si/SiO2 and thin NaCl layers. The hydrogen-catalyzed cyclodehydrogenation reaction reported here leads towards the integration of graphene derivatives in optoelectronic devices as well as developing the field of on-surface synthesis by means of catalytic transformations. It also inspires merging of atomically shaped graphene-based nanostructures with low-dimensional inorganic units into functional devices.},
note = {On-surface synthesis of nanographenes proceeds differently on metals than on semiconductors or insulating substrates. Here, the authors perform substrate type-independent chemistry with atomic hydrogen acting as a catalyst in the intramolecular cyclodehydrogenation reaction of polyarenes, yielding atomically precise nanographenes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Atomically precise synthesis of graphene nanostructures on semiconductors and insulators has been a formidable challenge. In particular, the metallic substrates needed to catalyze cyclodehydrogenative planarization reactions limit subsequent applications that exploit the electronic and/or magnetic structure of graphene derivatives. Here, we introduce a protocol in which an on-surface reaction is initiated and carried out regardless of the substrate type. We demonstrate that, counterintuitively, atomic hydrogen can play the role of a catalyst in the cyclodehydrogenative planarization reaction. The high efficiency of the method is demonstrated by the nanographene synthesis on metallic Au, semiconducting TiO2, Ge:H, as well as on inert and insulating Si/SiO2 and thin NaCl layers. The hydrogen-catalyzed cyclodehydrogenation reaction reported here leads towards the integration of graphene derivatives in optoelectronic devices as well as developing the field of on-surface synthesis by means of catalytic transformations. It also inspires merging of atomically shaped graphene-based nanostructures with low-dimensional inorganic units into functional devices. |
| 196. | | Ying Xie, Yue Li, Zhisheng Peng, Chengyu Wang, Zanlin Qiu, Xinyi Cai, Tinglu Song, Jia Si, Xiaoxu Zhao, Liu Qian, Ziqiang Zhao, Jin Zhang Nano-seeding catalysts for high-density arrays of horizontally aligned carbon nanotubes with wafer-scale uniformity In: Nature Communications, vol. 16, no. 149, 2025, (Arrays of horizontally aligned carbon nanotubes are promising candidates for advanced integrated circuits. Here, the authors present a nano-seeding method that enables direct growth of such arrays on one-inch wafers with a density up to 140 tubes per μm.). @article{nokey,
title = {Nano-seeding catalysts for high-density arrays of horizontally aligned carbon nanotubes with wafer-scale uniformity},
author = {Ying Xie and Yue Li and Zhisheng Peng and Chengyu Wang and Zanlin Qiu and Xinyi Cai and Tinglu Song and Jia Si and Xiaoxu Zhao and Liu Qian and Ziqiang Zhao and Jin Zhang },
url = {https://www.nature.com/articles/s41467-024-55515-0.pdf},
doi = {10.1038/s41467-024-55515-0},
year = {2025},
date = {2025-01-02},
urldate = {2025-01-02},
journal = {Nature Communications},
volume = {16},
number = {149},
abstract = {In the realm of modern materials science, horizontally aligned carbon nanotube arrays stand as promising materials for the development of next-generation integrated circuits. However, their large-scale integration has been impeded by the constraints of current fabrication techniques, which struggle to achieve the necessary uniformity, density, and size control of carbon nanotube arrays. Overcoming this challenge necessitates a significant shift in fabrication approaches. Herein, we present a nano-seeding method that revolutionized the preparation of catalyst nanoparticles, crucial for carbon-nanotube-array synthesis. Our approach, underpinned by ion implantation and substrate processing, allows for precise control over catalyst formation. Further development of a vertical spraying chemical vapor deposition system homogenizes the gas flow and ensures the uniform growth of carbon nanotube arrays. This nano-seeding method culminates in the direct growth of one-inch carbon-nanotube-array wafers with the highest density of 140 tubes μm−1. The high density and uniformity of the as-prepared carbon-nanotube-array wafers are validated through an advanced high-throughput characterization technique. The electrical properties of high on-state current, high on/off ratio and low subthreshold swing are demonstrated in field-effect transistors based on the arrays. This study propels the scalability of carbon-nanotube-array fabrication for future carbon-based electronics.},
note = {Arrays of horizontally aligned carbon nanotubes are promising candidates for advanced integrated circuits. Here, the authors present a nano-seeding method that enables direct growth of such arrays on one-inch wafers with a density up to 140 tubes per μm.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In the realm of modern materials science, horizontally aligned carbon nanotube arrays stand as promising materials for the development of next-generation integrated circuits. However, their large-scale integration has been impeded by the constraints of current fabrication techniques, which struggle to achieve the necessary uniformity, density, and size control of carbon nanotube arrays. Overcoming this challenge necessitates a significant shift in fabrication approaches. Herein, we present a nano-seeding method that revolutionized the preparation of catalyst nanoparticles, crucial for carbon-nanotube-array synthesis. Our approach, underpinned by ion implantation and substrate processing, allows for precise control over catalyst formation. Further development of a vertical spraying chemical vapor deposition system homogenizes the gas flow and ensures the uniform growth of carbon nanotube arrays. This nano-seeding method culminates in the direct growth of one-inch carbon-nanotube-array wafers with the highest density of 140 tubes μm−1. The high density and uniformity of the as-prepared carbon-nanotube-array wafers are validated through an advanced high-throughput characterization technique. The electrical properties of high on-state current, high on/off ratio and low subthreshold swing are demonstrated in field-effect transistors based on the arrays. This study propels the scalability of carbon-nanotube-array fabrication for future carbon-based electronics. |
| 195. | | Jan Voigt, Miloš Baljozović, Kévin Martin, Christian Wäckerlin, Narcis Avarvari, Karl-Heinz Ernst An aperiodic chiral tiling by topological molecular self-assembly In: Nature Communications, vol. 16, no. 83, 2025, (Aperiodic tessellation is an important concept not only in art and mathematics, but also in materials science. Here the authors report on aperiodic tiling through two-dimensional molecular self-assembly on a surface involving dynamic chirality.). @article{nokey,
title = {An aperiodic chiral tiling by topological molecular self-assembly},
author = {Jan Voigt and Miloš Baljozović and Kévin Martin and Christian Wäckerlin and Narcis Avarvari and Karl-Heinz Ernst },
url = {https://www.nature.com/articles/s41467-024-55405-5.pdf},
doi = {10.1038/s41467-024-55405-5},
year = {2025},
date = {2025-01-02},
journal = {Nature Communications},
volume = {16},
number = {83},
abstract = {Studying the self-assembly of chiral molecules in two dimensions offers insights into the fundamentals of crystallization. Using scanning tunneling microscopy, we examine an uncommon aggregation of polyaromatic chiral molecules on a silver surface. Dense packing is achieved through a chiral triangular tiling of triads, with N and N ± 1 molecules at the edges. The triangles feature a random distribution of mirror-isomers, with a significant excess of one isomer. Chirality at the domain boundaries causes a lateral shift, producing three distinct topological defects where six triangles converge. These defects partially contribute to the formation of supramolecular spirals. The observation of different equal-density arrangements suggests that entropy maximization must play a crucial role. Despite the potential for regular patterns, all observed tiling is aperiodic. Differences from previously reported aperiodic molecular assemblies, such as Penrose tiling, are discussed. Our findings demonstrate that two-dimensional molecular self-assembly can be governed by topological constraints, leading to aperiodic tiling induced by intermolecular forces.},
note = {Aperiodic tessellation is an important concept not only in art and mathematics, but also in materials science. Here the authors report on aperiodic tiling through two-dimensional molecular self-assembly on a surface involving dynamic chirality.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Studying the self-assembly of chiral molecules in two dimensions offers insights into the fundamentals of crystallization. Using scanning tunneling microscopy, we examine an uncommon aggregation of polyaromatic chiral molecules on a silver surface. Dense packing is achieved through a chiral triangular tiling of triads, with N and N ± 1 molecules at the edges. The triangles feature a random distribution of mirror-isomers, with a significant excess of one isomer. Chirality at the domain boundaries causes a lateral shift, producing three distinct topological defects where six triangles converge. These defects partially contribute to the formation of supramolecular spirals. The observation of different equal-density arrangements suggests that entropy maximization must play a crucial role. Despite the potential for regular patterns, all observed tiling is aperiodic. Differences from previously reported aperiodic molecular assemblies, such as Penrose tiling, are discussed. Our findings demonstrate that two-dimensional molecular self-assembly can be governed by topological constraints, leading to aperiodic tiling induced by intermolecular forces. |
| 194. | | Shiyuan Liu, Ying Hong, Wang Hong, Yi Zheng, Xiaodan Yang, Xuemu Li, Zhuomin Zhang, Xiaodong Yan, Yao Shan, Weikang Lin, Zehua Peng, Xingqi Zhang, Xi Yao, Zuankai Wang, Zhengbao Yang Stress-eliminated liquid-phase fabrication of colloidal films above the critical crack thickness In: Nature Communications, vol. 15, no. 10136, 2024, (There is a critical crack thickness in the production of thin films. Here, the authors utilize the surface tension of the liquid to eliminate the stresses within the film and produce ceramic layers with a controllable thickness and shape.). @article{nokey,
title = {Stress-eliminated liquid-phase fabrication of colloidal films above the critical crack thickness},
author = {Shiyuan Liu and Ying Hong and Wang Hong and Yi Zheng and Xiaodan Yang and Xuemu Li and Zhuomin Zhang and Xiaodong Yan and Yao Shan and Weikang Lin and Zehua Peng and Xingqi Zhang and Xi Yao and Zuankai Wang and Zhengbao Yang },
url = {https://www.nature.com/articles/s41467-024-54412-w.pdf},
doi = {10.1038/s41467-024-54412-w},
year = {2024},
date = {2024-12-02},
urldate = {2024-12-02},
journal = {Nature Communications},
volume = {15},
number = {10136},
abstract = {The thickness of film materials is a critical factor influencing properties such as energy density, optical performance, and mechanical strength. However, the long-standing challenge of the intrinsic thermodynamic limit on maximum thickness often leads to detrimental cracking, compromising these desirable properties. In this study, we present an approach called the stress-eliminated liquid-phase fabrication (SELF) method. The SELF method eliminates the need for substrates to support the precursor solution used for film fabrication. We harness the intrinsic surface tension of the solution by confining it within specifically designed grids in a framework, forming suspended liquid bridges. This technique enables fabrication of crack-free ceramic films within a broad thickness range from 1 to 100 μm. Furthermore, the fabricated PZT films exhibit a high piezoelectric coefficient (d33) of 229 pC N−1. The customizable grids not only offer design freedom for film topologies but also facilitate the fabrication of diverse film arrays without the need for destructive cutting processes. Moreover, the freestanding nature of these films enhances their adaptability for MEMS processing, and the “capillary bridge” topology allows the PZT films to be used in ultrasound focusing transmitter, providing possibilities in the medical imaging.},
note = {There is a critical crack thickness in the production of thin films. Here, the authors utilize the surface tension of the liquid to eliminate the stresses within the film and produce ceramic layers with a controllable thickness and shape.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The thickness of film materials is a critical factor influencing properties such as energy density, optical performance, and mechanical strength. However, the long-standing challenge of the intrinsic thermodynamic limit on maximum thickness often leads to detrimental cracking, compromising these desirable properties. In this study, we present an approach called the stress-eliminated liquid-phase fabrication (SELF) method. The SELF method eliminates the need for substrates to support the precursor solution used for film fabrication. We harness the intrinsic surface tension of the solution by confining it within specifically designed grids in a framework, forming suspended liquid bridges. This technique enables fabrication of crack-free ceramic films within a broad thickness range from 1 to 100 μm. Furthermore, the fabricated PZT films exhibit a high piezoelectric coefficient (d33) of 229 pC N−1. The customizable grids not only offer design freedom for film topologies but also facilitate the fabrication of diverse film arrays without the need for destructive cutting processes. Moreover, the freestanding nature of these films enhances their adaptability for MEMS processing, and the “capillary bridge” topology allows the PZT films to be used in ultrasound focusing transmitter, providing possibilities in the medical imaging. |
| 193. | | Vahid Shahabadi, Benjamin Vennes, Ryan Schmedding, Andreas Zuend, Janine Mauzeroll, Steen B. Schougaard, Thomas C. Preston Quantifying surface tension of metastable aerosols via electrodeformation In: Nature Communications, vol. 15, no. 10457, 2024, (Atmospheric aerosols are often in metastable states. Here, the authors present a non-contact method for measuring the surface tension in single microdroplets using electrodeformation and Raman scattering, which enables precise measurement of such states.). @article{nokey,
title = {Quantifying surface tension of metastable aerosols via electrodeformation},
author = {Vahid Shahabadi and Benjamin Vennes and Ryan Schmedding and Andreas Zuend and Janine Mauzeroll and Steen B. Schougaard and Thomas C. Preston },
url = {https://www.nature.com/articles/s41467-024-54106-3.pdf},
doi = {10.1038/s41467-024-54106-3},
year = {2024},
date = {2024-12-02},
journal = {Nature Communications},
volume = {15},
number = {10457},
abstract = {Accurate surface tension measurements are key to understanding and predicting the behavior of atmospheric aerosols, particularly their formation, growth, and phase transitions. In Earth’s atmosphere, aerosols often exist in metastable states, such as being supercooled or supersaturated. Standard tensiometry instruments face challenges in accessing these states due to the large sample volumes they require and rapid phase changes near surfaces. We present an instrument that uses a strong electric field, nearing the dielectric strength of air, to deform aerosol microdroplets and measure surface tension in a contact-free, humidity-controlled environment. A dual-beam optical trap holds single microdroplets between two electrodes and excites Raman scattering. When a high voltage is applied, droplet deformations reach tens of nanometers. These small shape changes are precisely measured through the splitting of morphology-dependent resonances, seen as sharp peaks in Raman spectra. Our measurements cover water activities where droplets are supersaturated, a region with limited previous data, and show good agreement with existing data where comparisons are possible. Unlike prior levitation-based methods, this approach measures surface tension in systems with viscosities over 102 Pa s without relying on dynamic processes.},
note = {Atmospheric aerosols are often in metastable states. Here, the authors present a non-contact method for measuring the surface tension in single microdroplets using electrodeformation and Raman scattering, which enables precise measurement of such states.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Accurate surface tension measurements are key to understanding and predicting the behavior of atmospheric aerosols, particularly their formation, growth, and phase transitions. In Earth’s atmosphere, aerosols often exist in metastable states, such as being supercooled or supersaturated. Standard tensiometry instruments face challenges in accessing these states due to the large sample volumes they require and rapid phase changes near surfaces. We present an instrument that uses a strong electric field, nearing the dielectric strength of air, to deform aerosol microdroplets and measure surface tension in a contact-free, humidity-controlled environment. A dual-beam optical trap holds single microdroplets between two electrodes and excites Raman scattering. When a high voltage is applied, droplet deformations reach tens of nanometers. These small shape changes are precisely measured through the splitting of morphology-dependent resonances, seen as sharp peaks in Raman spectra. Our measurements cover water activities where droplets are supersaturated, a region with limited previous data, and show good agreement with existing data where comparisons are possible. Unlike prior levitation-based methods, this approach measures surface tension in systems with viscosities over 102 Pa s without relying on dynamic processes. |
| 192. | | Pieter J. Keenan, Rebecca M. Purkiss, Tillmann Klamroth, Peter A. Sloan, Kristina R. Rusimova
Measuring competing outcomes of a single-molecule reaction reveals classical Arrhenius chemical kinetics In: Nature Communications, vol. 15, no. 10322, 2024, (Controlling matter one molecule at a time is the ultimate goal of nanotechnology. Here, the authors show that it is possible to use a scanning tunnelling microscope to measure and influence the outcome of a single-molecule reaction with multiple competing outcomes.). @article{nokey,
title = {Measuring competing outcomes of a single-molecule reaction reveals classical Arrhenius chemical kinetics},
author = {Pieter J. Keenan and Rebecca M. Purkiss and Tillmann Klamroth and Peter A. Sloan and Kristina R. Rusimova
},
url = {https://www.nature.com/articles/s41467-024-54677-1.pdf},
doi = {10.1038/s41467-024-54677-1},
year = {2024},
date = {2024-11-28},
journal = {Nature Communications},
volume = {15},
number = {10322},
abstract = {Programming matter one molecule at a time is a long-standing goal in nanoscience. The atomic resolution of a scanning tunnelling microscope (STM) can give control over the probability of inducing single-outcome single-molecule reactions. Here we show it is possible to measure and influence the outcome of a single-molecule reaction with multiple competing outcomes. By precise injection of electrons from an STM tip, toluene molecules are induced to react with two outcomes: switching to an adjacent site or desorption. Within a voltage range set by the electronic structure of the molecule-surface system, we see that the branching ratio between these two outcomes is dependent on the excess energy the exciting electron carries. Using known values, ab-initio DFT calculations and empirical models, we conclude that this excess energy leads to a heating of a common intermediate physisorbed state and gives control over the two outcomes via their energy barriers and prefactors.},
note = {Controlling matter one molecule at a time is the ultimate goal of nanotechnology. Here, the authors show that it is possible to use a scanning tunnelling microscope to measure and influence the outcome of a single-molecule reaction with multiple competing outcomes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Programming matter one molecule at a time is a long-standing goal in nanoscience. The atomic resolution of a scanning tunnelling microscope (STM) can give control over the probability of inducing single-outcome single-molecule reactions. Here we show it is possible to measure and influence the outcome of a single-molecule reaction with multiple competing outcomes. By precise injection of electrons from an STM tip, toluene molecules are induced to react with two outcomes: switching to an adjacent site or desorption. Within a voltage range set by the electronic structure of the molecule-surface system, we see that the branching ratio between these two outcomes is dependent on the excess energy the exciting electron carries. Using known values, ab-initio DFT calculations and empirical models, we conclude that this excess energy leads to a heating of a common intermediate physisorbed state and gives control over the two outcomes via their energy barriers and prefactors. |
| 191. | | Tingting Hao, Huiqian Zhou, Panpan Gai, Zhaoliang Wang, Yuxin Guo, Han Lin, Wenting Wei, Zhiyong Guo Deep learning-assisted single-atom detection of copper ions by combining click chemistry and fast scan voltammetry In: Nature Communications, vol. 15, no. 10292, 2024, (Life activities are inextricably linked to the regulation of trace copper ions. Here, the authors report a deep learning-assisted electrochemical sensor for single-atom detection of copper ions based on click chemistry and fast scan voltammetry.). @article{nokey,
title = {Deep learning-assisted single-atom detection of copper ions by combining click chemistry and fast scan voltammetry},
author = {Tingting Hao and Huiqian Zhou and Panpan Gai and Zhaoliang Wang and Yuxin Guo and Han Lin and Wenting Wei and Zhiyong Guo },
url = {https://www.nature.com/articles/s41467-024-54743-8.pdf},
doi = {10.1038/s41467-024-54743-8},
year = {2024},
date = {2024-11-27},
journal = {Nature Communications},
volume = {15},
number = {10292},
abstract = {Cell ion channels, cell proliferation and metastasis, and many other life activities are inseparable from the regulation of trace or even single copper ion (Cu+ and/or Cu2+). In this work, an electrochemical sensor for sensitive quantitative detection of 0.4−4 amol L−1 copper ions is developed by adopting: (1) copper ions catalyzing the click-chemistry reaction to capture numerous signal units; (2) special adsorption assembly method of signal units to ensure signal generation efficiency; and (3) fast scan voltammetry at 400 V s−1 to enhance signal intensity. And then, the single-atom detection of copper ions is realized by constructing a multi-layer deep convolutional neural network model FSVNet to extract hidden features and signal information of fast scan voltammograms for 0.2 amol L−1 of copper ions. Here, we show a multiple signal amplification strategy based on functionalized nanomaterials and fast scan voltammetry, together with a deep learning method, which realizes the sensitive detection and even single-atom detection of copper ions.},
note = {Life activities are inextricably linked to the regulation of trace copper ions. Here, the authors report a deep learning-assisted electrochemical sensor for single-atom detection of copper ions based on click chemistry and fast scan voltammetry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cell ion channels, cell proliferation and metastasis, and many other life activities are inseparable from the regulation of trace or even single copper ion (Cu+ and/or Cu2+). In this work, an electrochemical sensor for sensitive quantitative detection of 0.4−4 amol L−1 copper ions is developed by adopting: (1) copper ions catalyzing the click-chemistry reaction to capture numerous signal units; (2) special adsorption assembly method of signal units to ensure signal generation efficiency; and (3) fast scan voltammetry at 400 V s−1 to enhance signal intensity. And then, the single-atom detection of copper ions is realized by constructing a multi-layer deep convolutional neural network model FSVNet to extract hidden features and signal information of fast scan voltammograms for 0.2 amol L−1 of copper ions. Here, we show a multiple signal amplification strategy based on functionalized nanomaterials and fast scan voltammetry, together with a deep learning method, which realizes the sensitive detection and even single-atom detection of copper ions. |
| 190. |  | Chao Chen, Yinglin Ma, Kunda Yao, Qingmin Ji, Wei Liu Enantioselective adsorption on chiral ceramics with medium entropy In: Nature Communications, vol. 15, no. 10105, 2024, (Chiral metals often suffer from limitations such as reduced selectivity caused by kink coalescence and atomic roughness. Here, the authors present chiral medium-entropy ceramics and overcome the trade-off between stability and enantioselectivity.
). @article{nokey,
title = {Enantioselective adsorption on chiral ceramics with medium entropy},
author = {Chao Chen and Yinglin Ma and Kunda Yao and Qingmin Ji and Wei Liu },
url = {https://www.nature.com/articles/s41467-024-54414-8.pdf},
doi = {10.1038/s41467-024-54414-8},
year = {2024},
date = {2024-11-21},
urldate = {2024-11-21},
journal = {Nature Communications},
volume = {15},
number = {10105},
abstract = {Chiral metal surfaces provide an environment for enantioselective adsorption in various processes such as asymmetric catalysis, chiral recognition, and separation. However, they often suffer from limitations such as reduced enantioselectivity caused by kink coalescence and atomic roughness. Here, we present an approach using medium-entropy ceramic (MEC), specifically (CrMoTa)Si2 with a C40 hexagonal crystal structure, which overcomes the trade-off between thermal stability and enantioselectivity. Experimental confirmation is provided by employing quartz crystal microbalance (QCM), where the electrode is coated with MEC films using non-reactive magnetron sputtering technology. The chiral nature is verified through transmission electron microscopy and circular dichroism. Density-functional theory (DFT) calculations show that the stability of MEC films is significantly higher than that of high-index Cu surfaces. Through a combination of high-throughput DFT calculations and theoretical modeling, we demonstrate the high enantioselectivity (42% e.e.) of the chiral MEC for serine, a prototype molecule for studying enantioselective adsorption. The QCM results show that the adsorption amount of L-serine is 1.58 times higher than that of D-serine within a concentration range of 0-60 mM. These findings demonstrate the potential application of MECs in chiral recognition.},
note = {Chiral metals often suffer from limitations such as reduced selectivity caused by kink coalescence and atomic roughness. Here, the authors present chiral medium-entropy ceramics and overcome the trade-off between stability and enantioselectivity.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chiral metal surfaces provide an environment for enantioselective adsorption in various processes such as asymmetric catalysis, chiral recognition, and separation. However, they often suffer from limitations such as reduced enantioselectivity caused by kink coalescence and atomic roughness. Here, we present an approach using medium-entropy ceramic (MEC), specifically (CrMoTa)Si2 with a C40 hexagonal crystal structure, which overcomes the trade-off between thermal stability and enantioselectivity. Experimental confirmation is provided by employing quartz crystal microbalance (QCM), where the electrode is coated with MEC films using non-reactive magnetron sputtering technology. The chiral nature is verified through transmission electron microscopy and circular dichroism. Density-functional theory (DFT) calculations show that the stability of MEC films is significantly higher than that of high-index Cu surfaces. Through a combination of high-throughput DFT calculations and theoretical modeling, we demonstrate the high enantioselectivity (42% e.e.) of the chiral MEC for serine, a prototype molecule for studying enantioselective adsorption. The QCM results show that the adsorption amount of L-serine is 1.58 times higher than that of D-serine within a concentration range of 0-60 mM. These findings demonstrate the potential application of MECs in chiral recognition. |
| 189. | | Xufan Li, Samuel Wyss, Emanuil Yanev, Qing-Jie Li, Shuang Wu, Yongwen Sun, Raymond R. Unocic, Joseph Stage, Matthew Strasbourg, Lucas M. Sassi, Yingxin Zhu, Ju Li, Yang Yang, James Hone, Nicholas Borys, P. James Schuck, Avetik R. Harutyunyan Width-dependent continuous growth of atomically thin quantum nanoribbons from nanoalloy seeds in chalcogen vapor In: Nature Communications, vol. 15, no. 10080, 2024, (Size control in quantum materials by direct growth is still difficult to achieve. Here, the authors present the width-dependent growth of single-layer nanoribbons of transition metal dichalcogenides from nanoalloy seeds, achieving strain-induced quantum emission with a purity of up to 90% for single photons.). @article{nokey,
title = {Width-dependent continuous growth of atomically thin quantum nanoribbons from nanoalloy seeds in chalcogen vapor},
author = {Xufan Li and Samuel Wyss and Emanuil Yanev and Qing-Jie Li and Shuang Wu and Yongwen Sun and Raymond R. Unocic and Joseph Stage and Matthew Strasbourg and Lucas M. Sassi and Yingxin Zhu and Ju Li and Yang Yang and James Hone and Nicholas Borys and P. James Schuck and Avetik R. Harutyunyan },
url = {https://www.nature.com/articles/s41467-024-54413-9.pdf},
doi = {10.1038/s41467-024-54413-9},
year = {2024},
date = {2024-11-21},
urldate = {2024-11-21},
journal = {Nature Communications},
volume = {15},
number = {10080},
abstract = {Nanoribbons (NRs) of atomic layer transition metal dichalcogenides (TMDs) can boost the rapidly emerging field of quantum materials owing to their width-dependent phases and electronic properties. However, the controllable downscaling of width by direct growth and the underlying mechanism remain elusive. Here, we demonstrate the vapor-liquid-solid growth of single crystal of single layer NRs of a series of TMDs (MeX2: Me = Mo, W; X = S, Se) under chalcogen vapor atmosphere, seeded by pre-deposited and respective transition metal-alloyed nanoparticles that also control the NR width. We find linear dependence of growth rate on supersaturation, known as a criterion for continues growth mechanism, which decreases with decreasing of NR width driven by the Gibbs-Thomson effect. The NRs show width-dependent photoluminescence and strain-induced quantum emission signatures with up to ≈ 90% purity of single photons. We propose the path and underlying mechanism for width-controllable growth of TMD NRs for applications in quantum optoelectronics.},
note = {Size control in quantum materials by direct growth is still difficult to achieve. Here, the authors present the width-dependent growth of single-layer nanoribbons of transition metal dichalcogenides from nanoalloy seeds, achieving strain-induced quantum emission with a purity of up to 90% for single photons.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanoribbons (NRs) of atomic layer transition metal dichalcogenides (TMDs) can boost the rapidly emerging field of quantum materials owing to their width-dependent phases and electronic properties. However, the controllable downscaling of width by direct growth and the underlying mechanism remain elusive. Here, we demonstrate the vapor-liquid-solid growth of single crystal of single layer NRs of a series of TMDs (MeX2: Me = Mo, W; X = S, Se) under chalcogen vapor atmosphere, seeded by pre-deposited and respective transition metal-alloyed nanoparticles that also control the NR width. We find linear dependence of growth rate on supersaturation, known as a criterion for continues growth mechanism, which decreases with decreasing of NR width driven by the Gibbs-Thomson effect. The NRs show width-dependent photoluminescence and strain-induced quantum emission signatures with up to ≈ 90% purity of single photons. We propose the path and underlying mechanism for width-controllable growth of TMD NRs for applications in quantum optoelectronics. |
| 188. | | Xiaoli He, Hongri Gu, Yanmei Ma, Yuhang Cai, Huaide Jiang, Yi Zhang, Hanhan Xie, Ming Yang, Xinjian Fan, Liang Guo, Zhan Yang, Chengzhi Hu Light patterning semiconductor nanoparticles by modulating surface charges In: Nature Communications, vol. 15, no. 9843, 2024, (The integration of colloidal nanoparticles into microdevices is essential for several advanced technologies. Here, the authors have developed a scalable method of UV-triggered surface charge modulation for the rapid surface patterning with semiconductor nanoparticles.). @article{nokey,
title = {Light patterning semiconductor nanoparticles by modulating surface charges},
author = {Xiaoli He and Hongri Gu and Yanmei Ma and Yuhang Cai and Huaide Jiang and Yi Zhang and Hanhan Xie and Ming Yang and Xinjian Fan and Liang Guo and Zhan Yang and Chengzhi Hu },
url = {https://www.nature.com/articles/s41467-024-53926-7.pdf},
doi = {10.1038/s41467-024-53926-7},
year = {2024},
date = {2024-11-13},
journal = {Nature Communications},
volume = {15},
number = {9843},
abstract = {Optical patterning of colloidal particles is a scalable and cost-effective approach for creating multiscale functional structures. Existing methods often use high-intensity light sources and customized optical setups, making them less feasible for large-scale microfabrication processes. Here, we report an optical patterning method for semiconductor nanoparticles by light-triggered modulation of their surface charge. Rather than using light as the primary energy source, this method utilizes UV-induced cleavage of surface ligands to modify surface charges, thereby facilitating the self-assembly of nanoparticles on a charged substrate via electrostatic interactions. By using citrate-treated ZnO nanoparticles, uniform ZnO patterns with variable thicknesses can be achieved. These multilayered ZnO patterns are fabricated into a UV detector with an on/off ratio exceeding 10^4. Our results demonstrate a simple yet effective way to pattern semiconductor nanoparticles, facilitating the large-scale integration of functional nanomaterials into emerging flexible and robotic microdevices.},
note = {The integration of colloidal nanoparticles into microdevices is essential for several advanced technologies. Here, the authors have developed a scalable method of UV-triggered surface charge modulation for the rapid surface patterning with semiconductor nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Optical patterning of colloidal particles is a scalable and cost-effective approach for creating multiscale functional structures. Existing methods often use high-intensity light sources and customized optical setups, making them less feasible for large-scale microfabrication processes. Here, we report an optical patterning method for semiconductor nanoparticles by light-triggered modulation of their surface charge. Rather than using light as the primary energy source, this method utilizes UV-induced cleavage of surface ligands to modify surface charges, thereby facilitating the self-assembly of nanoparticles on a charged substrate via electrostatic interactions. By using citrate-treated ZnO nanoparticles, uniform ZnO patterns with variable thicknesses can be achieved. These multilayered ZnO patterns are fabricated into a UV detector with an on/off ratio exceeding 10^4. Our results demonstrate a simple yet effective way to pattern semiconductor nanoparticles, facilitating the large-scale integration of functional nanomaterials into emerging flexible and robotic microdevices. |
| 187. | | Wencong Zhang, Fan Li, Yi Li, Anran Song, Kun Yang, Dongchang Wu, Wen Shang, Zhenpeng Yao, Wenpei Gao, Tao Deng, Jianbo Wu The role of surface substitution in the atomic disorder-to-order phase transition in multi-component core–shell structures In: Nature Communications, vol. 15, no. 9762, 2024, (Understanding atomic-level ordering in multi-component systems is a challenge. Here, the authors investigate atom diffusion in cubic Pd@Pt-Co nanoparticles and find that substitution of Pd into (Pt, Pd)-Co lowers the phase transition temperature.). @article{nokey,
title = {The role of surface substitution in the atomic disorder-to-order phase transition in multi-component core–shell structures},
author = {Wencong Zhang and Fan Li and Yi Li and Anran Song and Kun Yang and Dongchang Wu and Wen Shang and Zhenpeng Yao and Wenpei Gao and Tao Deng and Jianbo Wu },
url = {https://www.nature.com/articles/s41467-024-54104-5.pdf},
doi = {10.1038/s41467-024-54104-5},
year = {2024},
date = {2024-11-11},
journal = {Nature Communications},
volume = {15},
number = {9762},
abstract = {Intermetallic phases with atomic ordering are highly active and stable in catalysts. However, understanding the atomistic mechanisms of disorder-to-order phase transition, particularly in multi-component systems, remains challenging. Here, we investigate the atom diffusion and phase transition within Pd@Pt-Co cubic nanoparticles during annealing, using in-situ electron microscopy and ex-situ atomic resolution element analysis. We reveal that initial outward diffusing Pd partially substitutes Pt, forming a (Pt, Pd)-Co ternary system in the surface region, enabling the phase transition at a low temperature of 400 °C, followed by shape-preserved inward propagation of the ordered phase. At higher temperatures, excessive interdiffusion across the interface changes the stoichiometric ratio, diminishing the atomic ordering, leading to obvious change in morphology. Calculations indicate that the Pd-substitute in (Pt, Pd)-Co system leads to a significantly lower phase transition temperature compared to that of Pt-Co alloy and thus a lower activation energy for atomic diffusion. These insights into atomistic behavior are crucial for future design of multi-component systems.},
note = {Understanding atomic-level ordering in multi-component systems is a challenge. Here, the authors investigate atom diffusion in cubic Pd@Pt-Co nanoparticles and find that substitution of Pd into (Pt, Pd)-Co lowers the phase transition temperature.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Intermetallic phases with atomic ordering are highly active and stable in catalysts. However, understanding the atomistic mechanisms of disorder-to-order phase transition, particularly in multi-component systems, remains challenging. Here, we investigate the atom diffusion and phase transition within Pd@Pt-Co cubic nanoparticles during annealing, using in-situ electron microscopy and ex-situ atomic resolution element analysis. We reveal that initial outward diffusing Pd partially substitutes Pt, forming a (Pt, Pd)-Co ternary system in the surface region, enabling the phase transition at a low temperature of 400 °C, followed by shape-preserved inward propagation of the ordered phase. At higher temperatures, excessive interdiffusion across the interface changes the stoichiometric ratio, diminishing the atomic ordering, leading to obvious change in morphology. Calculations indicate that the Pd-substitute in (Pt, Pd)-Co system leads to a significantly lower phase transition temperature compared to that of Pt-Co alloy and thus a lower activation energy for atomic diffusion. These insights into atomistic behavior are crucial for future design of multi-component systems. |
| 186. | | Niklas Friedrich, Anna Rosławska, Xabier Arrieta, Katharina Kaiser, Michelangelo Romeo, Eric Le Moal, Fabrice Scheurer, Javier Aizpurua, Andrei G. Borisov, Tomáš Neuman, Guillaume Schull Fluorescence from a single-molecule probe directly attached to a plasmonic STM tip In: Nature Communications, vol. 15, no. 9733, 2024, (Scanning tunneling microscopy (STM) gives access to the atomic-scale properties of matter. Here, the authors showcase the fluorescent functionalization of an STM tip using a single molecule in direct metal contact, permitting the local electrostatic and -dynamic environment to be probed.). @article{nokey,
title = {Fluorescence from a single-molecule probe directly attached to a plasmonic STM tip},
author = {Niklas Friedrich and Anna Rosławska and Xabier Arrieta and Katharina Kaiser and Michelangelo Romeo and Eric Le Moal and Fabrice Scheurer and Javier Aizpurua and Andrei G. Borisov and Tomáš Neuman and Guillaume Schull },
url = {https://www.nature.com/articles/s41467-024-53707-2.pdf},
doi = {10.1038/s41467-024-53707-2},
year = {2024},
date = {2024-11-10},
journal = {Nature Communications},
volume = {15},
number = {9733},
abstract = {The scanning tunneling microscope (STM) provides access to atomic-scale properties of a conductive sample. While single-molecule tip functionalization has become a standard procedure, fluorescent molecular probes remained absent from the available tool set. Here, the plasmonic tip of an STM is functionalized with a single fluorescent molecule and is scanned on a plasmonic substrate. The tunneling current flowing through the tip-molecule-substrate junction generates a narrow-line emission of light corresponding to the fluorescence of the negatively charged molecule suspended at the apex of the tip, i.e., the emission of the excited molecular anion. The fluorescence of this molecular probe is recorded for tip-substrate nanocavities featuring different plasmonic resonances, for different tip-substrate distances and applied bias voltages, and on different substrates. We demonstrate that the width of the emission peak can be used as a probe of the exciton-plasmon coupling strength and that the energy of the emitted photons is governed by the molecule interactions with its environment. Additionally, we theoretically elucidate why the direct contact of the suspended molecule with the metallic tip does not totally quench the radiative emission of the molecule.},
note = {Scanning tunneling microscopy (STM) gives access to the atomic-scale properties of matter. Here, the authors showcase the fluorescent functionalization of an STM tip using a single molecule in direct metal contact, permitting the local electrostatic and -dynamic environment to be probed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The scanning tunneling microscope (STM) provides access to atomic-scale properties of a conductive sample. While single-molecule tip functionalization has become a standard procedure, fluorescent molecular probes remained absent from the available tool set. Here, the plasmonic tip of an STM is functionalized with a single fluorescent molecule and is scanned on a plasmonic substrate. The tunneling current flowing through the tip-molecule-substrate junction generates a narrow-line emission of light corresponding to the fluorescence of the negatively charged molecule suspended at the apex of the tip, i.e., the emission of the excited molecular anion. The fluorescence of this molecular probe is recorded for tip-substrate nanocavities featuring different plasmonic resonances, for different tip-substrate distances and applied bias voltages, and on different substrates. We demonstrate that the width of the emission peak can be used as a probe of the exciton-plasmon coupling strength and that the energy of the emitted photons is governed by the molecule interactions with its environment. Additionally, we theoretically elucidate why the direct contact of the suspended molecule with the metallic tip does not totally quench the radiative emission of the molecule. |
| 185. | | Maxim Fatkullin, Dmitry Cheshev, Andrey Averkiev, Alina Gorbunova, Gennadiy Murastov, Jianxi Liu, Pavel Postnikov, Chong Cheng, Raul D. Rodriguez, Evgeniya Sheremet Photochemistry dominates over photothermal effects in the laser-induced reduction of graphene oxide by visible light In: Nature Communications, vol. 15, no. 9711, 2024, (Laser reduction of graphene oxide is generally assumed to be primarily driven by heat. Here the authors show that light-triggered chemical reactions dominate over photothermal effects.). @article{nokey,
title = {Photochemistry dominates over photothermal effects in the laser-induced reduction of graphene oxide by visible light},
author = {Maxim Fatkullin and Dmitry Cheshev and Andrey Averkiev and Alina Gorbunova and Gennadiy Murastov and Jianxi Liu and Pavel Postnikov and Chong Cheng and Raul D. Rodriguez and Evgeniya Sheremet },
url = {https://www.nature.com/articles/s41467-024-53503-y.pdf},
doi = {10.1038/s41467-024-53503-y},
year = {2024},
date = {2024-11-09},
journal = {Nature Communications},
volume = {15},
number = {9711},
abstract = {Graphene oxide (GO) possesses specific properties that are revolutionizing materials science, with applications extending from flexible electronics to advanced nanotechnology. A key method for harnessing GO’s potential is its laser-induced reduction, yet the exact mechanisms — photothermal versus photochemical effects — remain unclear. Herein, we discover the dominant role of photochemical reactions in the laser reduction of GO under visible light, challenging the prevailing assumption that photothermal effects are dominant. Employing a combination of Raman thermometry, X-ray photoelectron and photoluminescence spectroscopies, and electrical atomic force microscopy, we quantify the temperature and map the reduction process across micro and nano scales. Our findings demonstrate that the photochemical cleavage of oxygen-containing groups below a reduction threshold temperature is a decisive factor in GO reduction, leading to distinct characteristics that cannot be replicated by heating alone. This work clarifies the fundamental mechanisms of GO transformation under visible laser irradiation, highlighting the dominant role of photochemical processes. Distinguishing these subtleties enables the development of laser-reduced GO platforms for graphene-based applications compatible with industrial scales. We illustrate this potential by encoding information on GO surfaces as optical storage, allowing us to write binary sequences in long-term memory encoding invisible even through an optical microscope.},
note = {Laser reduction of graphene oxide is generally assumed to be primarily driven by heat. Here the authors show that light-triggered chemical reactions dominate over photothermal effects.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Graphene oxide (GO) possesses specific properties that are revolutionizing materials science, with applications extending from flexible electronics to advanced nanotechnology. A key method for harnessing GO’s potential is its laser-induced reduction, yet the exact mechanisms — photothermal versus photochemical effects — remain unclear. Herein, we discover the dominant role of photochemical reactions in the laser reduction of GO under visible light, challenging the prevailing assumption that photothermal effects are dominant. Employing a combination of Raman thermometry, X-ray photoelectron and photoluminescence spectroscopies, and electrical atomic force microscopy, we quantify the temperature and map the reduction process across micro and nano scales. Our findings demonstrate that the photochemical cleavage of oxygen-containing groups below a reduction threshold temperature is a decisive factor in GO reduction, leading to distinct characteristics that cannot be replicated by heating alone. This work clarifies the fundamental mechanisms of GO transformation under visible laser irradiation, highlighting the dominant role of photochemical processes. Distinguishing these subtleties enables the development of laser-reduced GO platforms for graphene-based applications compatible with industrial scales. We illustrate this potential by encoding information on GO surfaces as optical storage, allowing us to write binary sequences in long-term memory encoding invisible even through an optical microscope. |
| 184. | | Márkó Grabarics, Benjamín Mallada, Shayan Edalatmanesh, Alejandro Jiménez-Martín, Martin Pykal, Martin Ondráček, Petra Kührová, Weston B. Struwe, Pavel Banáš, Stephan Rauschenbach, Pavel Jelínek, Bruno de la Torre Atomically resolved imaging of the conformations and adsorption geometries of individual β-cyclodextrins with non-contact AFM In: Nature Communications, vol. 15, no. 9482, 2024, (Glycans are structurally complex biomolecules and very challenging to analyse. Here the authors show atomically resolved imaging of β-cyclodextrins with non-contact atomic force microscopy, revealing the structure of individual glycans with atomic detail.). @article{nokey,
title = {Atomically resolved imaging of the conformations and adsorption geometries of individual β-cyclodextrins with non-contact AFM},
author = {Márkó Grabarics and Benjamín Mallada and Shayan Edalatmanesh and Alejandro Jiménez-Martín and Martin Pykal and Martin Ondráček and Petra Kührová and Weston B. Struwe and Pavel Banáš and Stephan Rauschenbach and Pavel Jelínek and Bruno de la Torre },
url = {https://www.nature.com/articles/s41467-024-53555-0.pdf},
doi = {10.1038/s41467-024-53555-0},
year = {2024},
date = {2024-11-02},
journal = {Nature Communications},
volume = {15},
number = {9482},
abstract = {Glycans, consisting of covalently linked sugar units, are a major class of biopolymers essential to all known living organisms. To better understand their biological functions and further applications in fields from biomedicine to materials science, detailed knowledge of their structure is essential. However, due to the extraordinary complexity and conformational flexibility of glycans, state-of-the-art glycan analysis methods often fail to provide structural information with atomic precision. Here, we combine electrospray deposition in ultra-high vacuum with non-contact atomic force microscopy and theoretical calculations to unravel the structure of β-cyclodextrin, a cyclic glucose oligomer, with atomic-scale detail. Our results, established on the single-molecule level, reveal the different adsorption geometries and conformations of β-cyclodextrin. The position of individual hydroxy groups and the location of the stabilizing intramolecular H-bonds are deduced from atomically resolved images, enabling the unambiguous assignment of the molecular structure and demonstrating the potential of the method for glycan analysis.},
note = {Glycans are structurally complex biomolecules and very challenging to analyse. Here the authors show atomically resolved imaging of β-cyclodextrins with non-contact atomic force microscopy, revealing the structure of individual glycans with atomic detail.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Glycans, consisting of covalently linked sugar units, are a major class of biopolymers essential to all known living organisms. To better understand their biological functions and further applications in fields from biomedicine to materials science, detailed knowledge of their structure is essential. However, due to the extraordinary complexity and conformational flexibility of glycans, state-of-the-art glycan analysis methods often fail to provide structural information with atomic precision. Here, we combine electrospray deposition in ultra-high vacuum with non-contact atomic force microscopy and theoretical calculations to unravel the structure of β-cyclodextrin, a cyclic glucose oligomer, with atomic-scale detail. Our results, established on the single-molecule level, reveal the different adsorption geometries and conformations of β-cyclodextrin. The position of individual hydroxy groups and the location of the stabilizing intramolecular H-bonds are deduced from atomically resolved images, enabling the unambiguous assignment of the molecular structure and demonstrating the potential of the method for glycan analysis. |
| 183. | | Xiali Lv, Yu Tian, Fengxia Wu, Xiaoxi Luan, Fenghua Li, Zhili Shen, Guobao Xu, Kun Liu, Wenxin Niu Chiral plasmonic-dielectric coupling enables strong near-infrared chiroptical responses from helicoidal core-shell nanoparticles In: Nature Communications, vol. 15, no. 9234, 2024, (The rational design of chiroplasmonic nanomaterials with near-infrared (NIR) responses remains a challenge. Here, the authors propose a strategy utilizing chiral plasmonic-dielectric coupling to achieve strong chiroptical responses in the NIR region.). @article{nokey,
title = {Chiral plasmonic-dielectric coupling enables strong near-infrared chiroptical responses from helicoidal core-shell nanoparticles},
author = {Xiali Lv and Yu Tian and Fengxia Wu and Xiaoxi Luan and Fenghua Li and Zhili Shen and Guobao Xu and Kun Liu and Wenxin Niu },
url = {https://www.nature.com/articles/s41467-024-53705-4.pdf},
doi = {10.1038/s41467-024-53705-4},
year = {2024},
date = {2024-10-25},
urldate = {2024-10-25},
journal = {Nature Communications},
volume = {15},
number = {9234},
abstract = {Helicoid plasmonic nanoparticles with intrinsic chirality are an emerging class of artificial chiral materials with tailorable properties. The ability to extend their chiroplasmonic responses to the near-infrared (NIR) range is critically important for biomedical and nanophotonic applications, yet the rational design of such materials remains challenging. Herein, a strategy employing chiral plasmon-dielectric coupling is proposed to manipulate the chiroptical responses into the NIR region with high optical anisotropy. Through this strategy, the helicoid Au@Cu2O nanoparticles with structural chirality are designed and synthesized with tunable and enriched NIR chiroptical responses. Specially, a high optical anisotropy (g-factor) with a value of 0.35 is achieved in the NIR region, and multi-band chiroptical behaviors are observed. Spectral and electromagnetic simulations elucidate that strong coupling between chiroplasmonic core and chiral dielectric shell with high refractive index contributes to the rich and strong chiroptical responses, which are related to the interplay between various emerged and enhanced electric and magnetic multipolar resonance modes proved by multipole expansion analysis. Moreover, the helicoid Au@Cu2O nanoparticles display greater polarization rotation capability than the helicoid Au nanoparticles. This work offers mechanistic insights into chiral plasmon-dielectric coupling and suggests a general approach of creating NIR chiroplasmonic materials.},
note = {The rational design of chiroplasmonic nanomaterials with near-infrared (NIR) responses remains a challenge. Here, the authors propose a strategy utilizing chiral plasmonic-dielectric coupling to achieve strong chiroptical responses in the NIR region.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Helicoid plasmonic nanoparticles with intrinsic chirality are an emerging class of artificial chiral materials with tailorable properties. The ability to extend their chiroplasmonic responses to the near-infrared (NIR) range is critically important for biomedical and nanophotonic applications, yet the rational design of such materials remains challenging. Herein, a strategy employing chiral plasmon-dielectric coupling is proposed to manipulate the chiroptical responses into the NIR region with high optical anisotropy. Through this strategy, the helicoid Au@Cu2O nanoparticles with structural chirality are designed and synthesized with tunable and enriched NIR chiroptical responses. Specially, a high optical anisotropy (g-factor) with a value of 0.35 is achieved in the NIR region, and multi-band chiroptical behaviors are observed. Spectral and electromagnetic simulations elucidate that strong coupling between chiroplasmonic core and chiral dielectric shell with high refractive index contributes to the rich and strong chiroptical responses, which are related to the interplay between various emerged and enhanced electric and magnetic multipolar resonance modes proved by multipole expansion analysis. Moreover, the helicoid Au@Cu2O nanoparticles display greater polarization rotation capability than the helicoid Au nanoparticles. This work offers mechanistic insights into chiral plasmon-dielectric coupling and suggests a general approach of creating NIR chiroplasmonic materials. |
| 182. | | Gyeongwon Kang, Shu Hu, Chenyang Guo, Rakesh Arul, Sarah M. Sibug-Torres, Jeremy J. Baumberg Design rules for catalysis in single-particle plasmonic nanogap reactors with precisely aligned molecular monolayers In: Nature Communications, vol. 15, pp. 9220, 2024, (Plasmonic nanostructures can drive light-driven catalytic reactions, but controlling reaction kinetics remains challenging. Here, the authors design plasmonic nanoreactors that enhance control of catalytic reactions, revealing distinct kinetics based on molecular configuration and monolayer placement.). @article{nokey,
title = {Design rules for catalysis in single-particle plasmonic nanogap reactors with precisely aligned molecular monolayers},
author = {Gyeongwon Kang and Shu Hu and Chenyang Guo and Rakesh Arul and Sarah M. Sibug-Torres and Jeremy J. Baumberg },
url = {https://www.nature.com/articles/s41467-024-53544-3.pdf},
doi = {10.1038/s41467-024-53544-3},
year = {2024},
date = {2024-10-25},
urldate = {2024-10-25},
journal = {Nature Communications},
volume = {15},
pages = {9220},
abstract = {Plasmonic nanostructures can both drive and interrogate light-driven catalytic reactions. Sensitive detection of reaction pathways is achieved by confining optical fields near the active surface. However, effective control of the reaction kinetics remains a challenge to utilize nanostructure constructs as efficient chemical reactors. Here we present a nanoreactor construct exhibiting high catalytic and optical efficiencies, based on a nanoparticle-on-mirror (NPoM) platform. We observe and track pathways of the Pd-catalysed C-C coupling reaction of molecules within a set of nanogaps presenting different chemical surfaces. Atomic monolayer coatings of Pd on the different Au facets enable tuning of the reaction kinetics of surface-bound molecules. Systematic analysis shows the catalytic efficiency of NPoM-based nanoreactors greatly improves on platforms based on aggregated nanoparticles. More importantly, we show Pd monolayers on the nanoparticle or on the mirror play significantly different roles in the surface reaction kinetics. Our data provides clear evidence for catalytic dependencies on molecular configuration in well-defined nanostructures. Such nanoreactor constructs therefore yield clearer design rules for plasmonic catalysis.},
note = {Plasmonic nanostructures can drive light-driven catalytic reactions, but controlling reaction kinetics remains challenging. Here, the authors design plasmonic nanoreactors that enhance control of catalytic reactions, revealing distinct kinetics based on molecular configuration and monolayer placement.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Plasmonic nanostructures can both drive and interrogate light-driven catalytic reactions. Sensitive detection of reaction pathways is achieved by confining optical fields near the active surface. However, effective control of the reaction kinetics remains a challenge to utilize nanostructure constructs as efficient chemical reactors. Here we present a nanoreactor construct exhibiting high catalytic and optical efficiencies, based on a nanoparticle-on-mirror (NPoM) platform. We observe and track pathways of the Pd-catalysed C-C coupling reaction of molecules within a set of nanogaps presenting different chemical surfaces. Atomic monolayer coatings of Pd on the different Au facets enable tuning of the reaction kinetics of surface-bound molecules. Systematic analysis shows the catalytic efficiency of NPoM-based nanoreactors greatly improves on platforms based on aggregated nanoparticles. More importantly, we show Pd monolayers on the nanoparticle or on the mirror play significantly different roles in the surface reaction kinetics. Our data provides clear evidence for catalytic dependencies on molecular configuration in well-defined nanostructures. Such nanoreactor constructs therefore yield clearer design rules for plasmonic catalysis. |
| 181. | | Miao Song, Dingri Zhang, Dan Leng, Jaewon Lee, Ziang Yang, Jiaxuan Chen, Dan Li, Lei Wang, Gang Zhou, Rui Yang, Kechao Zhou In situ atomic observations of aggregation growth and evolution of penta-twinned gold nanocrystals In: Nature Communications, vol. 15, no. 9217, 2024, (The mechanisms underlying the nonclassical growth of penta-twinned gold nanocrystals remain unclear. Here, the authors elucidate the atomic-level processes that drive size-dependent and twin configuration-related aggregation phenomena.
). @article{nokey,
title = {In situ atomic observations of aggregation growth and evolution of penta-twinned gold nanocrystals},
author = {Miao Song and Dingri Zhang and Dan Leng and Jaewon Lee and Ziang Yang and Jiaxuan Chen and Dan Li and Lei Wang and Gang Zhou and Rui Yang and Kechao Zhou },
url = {https://www.nature.com/articles/s41467-024-53501-0.pdf},
doi = {10.1038/s41467-024-53501-0},
year = {2024},
date = {2024-10-25},
journal = {Nature Communications},
volume = {15},
number = {9217},
abstract = {The twin boundaries and inherent lattice strain of five-fold twin (5-FT) structures offer a promising and innovative approach to tune nanocrystal configurations and properties, enriching nanomaterial performance. However, a comprehensive understanding of the nonclassical growth models governing 5-FT nanocrystals remains elusive, largely due to the constraints of their small thermodynamically stable size and complex twin configurations. Here, we conducted in situ investigations to elucidate the atomic-scale mechanisms driving size-dependent and twin configuration-related aggregation phenomena between 5-FT and other nanoparticles at the atomic scale. Our results reveal that surface diffusion significantly shapes the morphology of aggregated nanoparticles, promoting the symmetrical formation of 5-FT, especially in smaller nanoparticles. Moreover, the inherent structural characteristics of 5-FT mitigate the dominance of surface diffusion in its morphological evolution, retarding the aggregation evolution process and fostering intricate twin structures. These findings contribute to advancing our capacity to manipulate the configuration of twinned particles, enabling more predictable synthesis of functional nanomaterials for advanced engineering applications.},
note = {The mechanisms underlying the nonclassical growth of penta-twinned gold nanocrystals remain unclear. Here, the authors elucidate the atomic-level processes that drive size-dependent and twin configuration-related aggregation phenomena.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The twin boundaries and inherent lattice strain of five-fold twin (5-FT) structures offer a promising and innovative approach to tune nanocrystal configurations and properties, enriching nanomaterial performance. However, a comprehensive understanding of the nonclassical growth models governing 5-FT nanocrystals remains elusive, largely due to the constraints of their small thermodynamically stable size and complex twin configurations. Here, we conducted in situ investigations to elucidate the atomic-scale mechanisms driving size-dependent and twin configuration-related aggregation phenomena between 5-FT and other nanoparticles at the atomic scale. Our results reveal that surface diffusion significantly shapes the morphology of aggregated nanoparticles, promoting the symmetrical formation of 5-FT, especially in smaller nanoparticles. Moreover, the inherent structural characteristics of 5-FT mitigate the dominance of surface diffusion in its morphological evolution, retarding the aggregation evolution process and fostering intricate twin structures. These findings contribute to advancing our capacity to manipulate the configuration of twinned particles, enabling more predictable synthesis of functional nanomaterials for advanced engineering applications. |
| 180. | | Andrei Bylinkin, Sebastián Castilla, Tetiana M. Slipchenko, Kateryna Domina, Francesco Calavalle, Varun-Varma Pusapati, Marta Autore, Fèlix Casanova, Luis E. Hueso, Luis Martín-Moreno, Alexey Y. Nikitin, Frank H. L. Koppens, Rainer Hillenbrand On-chip phonon-enhanced IR near-field detection of molecular vibrations In: Nature Communications, vol. 15, no. 8907, 2024. @article{nokey,
title = {On-chip phonon-enhanced IR near-field detection of molecular vibrations},
author = {Andrei Bylinkin and Sebastián Castilla and Tetiana M. Slipchenko and Kateryna Domina and Francesco Calavalle and Varun-Varma Pusapati and Marta Autore and Fèlix Casanova and Luis E. Hueso and Luis Martín-Moreno and Alexey Y. Nikitin and Frank H. L. Koppens and Rainer Hillenbrand },
url = {https://www.nature.com/articles/s41467-024-53182-9.pdf},
doi = {10.1038/s41467-024-53182-9},
year = {2024},
date = {2024-10-16},
journal = {Nature Communications},
volume = {15},
number = {8907},
abstract = {Phonon polaritons – quasiparticles formed by strong coupling of infrared (IR) light with lattice vibrations in polar materials – can be utilized for surface-enhanced infrared absorption (SEIRA) spectroscopy and even for vibrational strong coupling with nanoscale amounts of molecules. Here, we introduce and demonstrate a compact on-chip phononic SEIRA spectroscopy platform, which is based on an h-BN/graphene/h-BN heterostructure on top of a metal split-gate creating a p-n junction in graphene. The metal split-gate concentrates the incident light and launches hyperbolic phonon polaritons (HPhPs) in the heterostructure, which serves simultaneously as SEIRA substrate and room-temperature infrared detector. When thin organic layers are deposited directly on top of the heterostructure, we observe a photocurrent encoding the layer’s molecular vibrational fingerprint, which is strongly enhanced compared to that observed in standard far-field absorption spectroscopy. A detailed theoretical analysis supports our results, further predicting an additional sensitivity enhancement as the molecular layers approach deep subwavelength scales. Future on-chip integration of infrared light sources such as quantum cascade lasers or even electrical generation of the HPhPs could lead to fully on-chip phononic SEIRA sensors for molecular and gas sensing.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Phonon polaritons – quasiparticles formed by strong coupling of infrared (IR) light with lattice vibrations in polar materials – can be utilized for surface-enhanced infrared absorption (SEIRA) spectroscopy and even for vibrational strong coupling with nanoscale amounts of molecules. Here, we introduce and demonstrate a compact on-chip phononic SEIRA spectroscopy platform, which is based on an h-BN/graphene/h-BN heterostructure on top of a metal split-gate creating a p-n junction in graphene. The metal split-gate concentrates the incident light and launches hyperbolic phonon polaritons (HPhPs) in the heterostructure, which serves simultaneously as SEIRA substrate and room-temperature infrared detector. When thin organic layers are deposited directly on top of the heterostructure, we observe a photocurrent encoding the layer’s molecular vibrational fingerprint, which is strongly enhanced compared to that observed in standard far-field absorption spectroscopy. A detailed theoretical analysis supports our results, further predicting an additional sensitivity enhancement as the molecular layers approach deep subwavelength scales. Future on-chip integration of infrared light sources such as quantum cascade lasers or even electrical generation of the HPhPs could lead to fully on-chip phononic SEIRA sensors for molecular and gas sensing. |
| 179. | | Cong Zhao, Jiazheng Diao, Zhao Liu, Jie Hao, Suhang He, Shaojia Li, Xingxing Li, Guangwu Li, Qiang Fu, Chuancheng Jia, Xuefeng Guo Electrical monitoring of single-event protonation dynamics at the solid-liquid interface and its regulation by external mechanical forces In: Nature Communications, vol. 15, no. 8835, 2024, (Direct detection of chemical reactions at solid-liquid interfaces is important for a more complete understanding of interfacial effects. Here, the authors study single-event interfacial protonation reaction dynamics by single-molecule junctions and thereby identify an interfacial cationic state.). @article{nokey,
title = {Electrical monitoring of single-event protonation dynamics at the solid-liquid interface and its regulation by external mechanical forces},
author = {Cong Zhao and Jiazheng Diao and Zhao Liu and Jie Hao and Suhang He and Shaojia Li and Xingxing Li and Guangwu Li and Qiang Fu and Chuancheng Jia and Xuefeng Guo },
url = {https://www.nature.com/articles/s41467-024-53179-4.pdf},
doi = {10.1038/s41467-024-53179-4},
year = {2024},
date = {2024-10-13},
urldate = {2024-10-13},
journal = {Nature Communications},
volume = {15},
number = {8835},
abstract = {Detecting chemical reaction dynamics at solid-liquid interfaces is important for understanding heterogeneous reactions. However, there is a lack of exploration of interface reaction dynamics from the single-molecule perspective, which can reveal the intrinsic reaction mechanism underlying ensemble experiments. Here, single-event protonation reaction dynamics at a solid-liquid interface are studied in-situ using single-molecule junctions. Molecules with amino terminal groups are used to construct single-molecule junctions. An interfacial cationic state present after protonation is discovered. Real-time electrical measurements are used to monitor the reversible reaction between protonated and deprotonated states, thereby revealing the interfacial reaction mechanism through dynamic analysis. The protonation reaction rate constant has a linear positive correlation with proton concentration, whereas the deprotonation reaction rate constant has a linear negative correlation. In addition, external mechanical forces can effectively regulate the protonation reaction process. This work provides a single-molecule perspective for exploring interface science, which will contribute to the development of heterogeneous catalysis and electrochemistry.},
note = {Direct detection of chemical reactions at solid-liquid interfaces is important for a more complete understanding of interfacial effects. Here, the authors study single-event interfacial protonation reaction dynamics by single-molecule junctions and thereby identify an interfacial cationic state.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Detecting chemical reaction dynamics at solid-liquid interfaces is important for understanding heterogeneous reactions. However, there is a lack of exploration of interface reaction dynamics from the single-molecule perspective, which can reveal the intrinsic reaction mechanism underlying ensemble experiments. Here, single-event protonation reaction dynamics at a solid-liquid interface are studied in-situ using single-molecule junctions. Molecules with amino terminal groups are used to construct single-molecule junctions. An interfacial cationic state present after protonation is discovered. Real-time electrical measurements are used to monitor the reversible reaction between protonated and deprotonated states, thereby revealing the interfacial reaction mechanism through dynamic analysis. The protonation reaction rate constant has a linear positive correlation with proton concentration, whereas the deprotonation reaction rate constant has a linear negative correlation. In addition, external mechanical forces can effectively regulate the protonation reaction process. This work provides a single-molecule perspective for exploring interface science, which will contribute to the development of heterogeneous catalysis and electrochemistry. |
| 178. | | Nicholas Drachman, Mathilde Lepoitevin, Hannah Szapary, Benjamin Wiener, William Maulbetsch, Derek Stein Nanopore ion sources deliver individual ions of amino acids and peptides directly into high vacuum In: Nature Communications, vol. 15, no. 7709, 2024, (Electrospray ionization loses most ions upon transfer into high vacuum in a mass spectrometer. Here, the authors present a nanopore ion source that emits ions directly into vacuum from aqueous solutions, achieving an ion transmission efficiency of over 90%.). @article{nokey,
title = {Nanopore ion sources deliver individual ions of amino acids and peptides directly into high vacuum},
author = {Nicholas Drachman and Mathilde Lepoitevin and Hannah Szapary and Benjamin Wiener and William Maulbetsch and Derek Stein },
url = {https://www.nature.com/articles/s41467-024-51455-x.pdf},
doi = {10.1038/s41467-024-51455-x},
year = {2024},
date = {2024-09-05},
urldate = {2024-09-05},
journal = {Nature Communications},
volume = {15},
number = {7709},
abstract = {Electrospray ionization is widely used to generate vapor phase ions for analysis by mass spectrometry in proteomics research. However, only a small fraction of the analyte enters the mass spectrometer due to losses that are fundamentally linked to the use of a background gas to stimulate the generation of ions from electrosprayed droplets. Here we report a nanopore ion source that delivers ions directly into high vacuum from aqueous solutions. The ion source comprises a pulled quartz pipette with a sub-100 nm opening. Ions escape an electrified meniscus by ion evaporation and travel along collisionless trajectories to the ion detector. We measure mass spectra of 16 different amino acid ions, post-translationally modified variants of glutathione, and the peptide angiotensin II, showing that these analytes can be emitted as desolvated ions. The emitted current is composed of ions rather than charged droplets, and more than 90% of the current can be recovered in a distant collector.},
note = {Electrospray ionization loses most ions upon transfer into high vacuum in a mass spectrometer. Here, the authors present a nanopore ion source that emits ions directly into vacuum from aqueous solutions, achieving an ion transmission efficiency of over 90%.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Electrospray ionization is widely used to generate vapor phase ions for analysis by mass spectrometry in proteomics research. However, only a small fraction of the analyte enters the mass spectrometer due to losses that are fundamentally linked to the use of a background gas to stimulate the generation of ions from electrosprayed droplets. Here we report a nanopore ion source that delivers ions directly into high vacuum from aqueous solutions. The ion source comprises a pulled quartz pipette with a sub-100 nm opening. Ions escape an electrified meniscus by ion evaporation and travel along collisionless trajectories to the ion detector. We measure mass spectra of 16 different amino acid ions, post-translationally modified variants of glutathione, and the peptide angiotensin II, showing that these analytes can be emitted as desolvated ions. The emitted current is composed of ions rather than charged droplets, and more than 90% of the current can be recovered in a distant collector. |
| 177. | | Yao Zhang, Zezhou Li, Xing Tong, Zhiheng Xie, Siwei Huang, Yue-E Zhang, Hai-Bo Ke, Wei-Hua Wang, Jihan Zhou Three-dimensional atomic insights into the metal-oxide interface in Zr-ZrO2 nanoparticles In: Nature Communications, vol. 15, no. 7624, 2024, (A detailed understanding of metal-oxide interfaces is essential for uncovering their intrinsic properties. Here, the authors investigate the 3D atomic structure of metal-oxide interfaces in Zr-ZrO2 nanoparticles using atomic-resolution electron tomography.
). @article{nokey,
title = {Three-dimensional atomic insights into the metal-oxide interface in Zr-ZrO2 nanoparticles},
author = {Yao Zhang and Zezhou Li and Xing Tong and Zhiheng Xie and Siwei Huang and Yue-E Zhang and Hai-Bo Ke and Wei-Hua Wang and Jihan Zhou },
url = {https://www.nature.com/articles/s41467-024-52026-w.pdf},
doi = {10.1038/s41467-024-52026-w},
year = {2024},
date = {2024-09-02},
journal = {Nature Communications},
volume = {15},
number = {7624},
abstract = {Metal-oxide interfaces with poor coherency have specific properties comparing to bulk materials and offer broad applications in heterogeneous catalysis, battery, and electronics. However, current understanding of the three-dimensional (3D) atomic metal-oxide interfaces remains limited because of their inherent structural complexity and the limitations of conventional two-dimensional imaging techniques. Here, we determine the 3D atomic structure of metal-oxide interfaces in zirconium-zirconia nanoparticles using atomic-resolution electron tomography. We quantitatively analyze the atomic concentration and the degree of oxidation, and find the coherency and translational symmetry of the interfaces are broken. Atoms at the interface have low structural ordering, low coordination, and elongated bond length. Moreover, we observe porous structures such as Zr vacancies and nano-pores, and investigate their distribution. Our findings provide a clear 3D atomic picture of metal-oxide interface with direct experimental evidence. We anticipate this work could encourage future studies on fundamental problems of oxides, such as interfacial structures in semiconductor and atomic motion during oxidation process.},
note = {A detailed understanding of metal-oxide interfaces is essential for uncovering their intrinsic properties. Here, the authors investigate the 3D atomic structure of metal-oxide interfaces in Zr-ZrO2 nanoparticles using atomic-resolution electron tomography.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Metal-oxide interfaces with poor coherency have specific properties comparing to bulk materials and offer broad applications in heterogeneous catalysis, battery, and electronics. However, current understanding of the three-dimensional (3D) atomic metal-oxide interfaces remains limited because of their inherent structural complexity and the limitations of conventional two-dimensional imaging techniques. Here, we determine the 3D atomic structure of metal-oxide interfaces in zirconium-zirconia nanoparticles using atomic-resolution electron tomography. We quantitatively analyze the atomic concentration and the degree of oxidation, and find the coherency and translational symmetry of the interfaces are broken. Atoms at the interface have low structural ordering, low coordination, and elongated bond length. Moreover, we observe porous structures such as Zr vacancies and nano-pores, and investigate their distribution. Our findings provide a clear 3D atomic picture of metal-oxide interface with direct experimental evidence. We anticipate this work could encourage future studies on fundamental problems of oxides, such as interfacial structures in semiconductor and atomic motion during oxidation process. |
| 176. | | Xiao Han, Yanan Zhou, Xiaolin Tai, Geng Wu, Cai Chen, Xun Hong, Lei Tong, Fangfang Xu, Hai-Wei Liang, Yue Lin In-situ atomic tracking of intermetallic compound formation during thermal annealing In: Nature Communications, vol. 15, no. 7200, 2024, (The mechanisms of intermetallic compound (IMC) formation during annealing are poorly understood. Here, the authors identify five stages of PtFe-IMC formation at the atomic level using in-situ STEM, emphasizing the role of high temperature in achieving the targeted stoichiometric ratio.). @article{nokey,
title = {In-situ atomic tracking of intermetallic compound formation during thermal annealing},
author = {Xiao Han and Yanan Zhou and Xiaolin Tai and Geng Wu and Cai Chen and Xun Hong and Lei Tong and Fangfang Xu and Hai-Wei Liang and Yue Lin },
url = {https://www.nature.com/articles/s41467-024-51541-0.pdf},
doi = {10.1038/s41467-024-51541-0},
year = {2024},
date = {2024-08-22},
journal = {Nature Communications},
volume = {15},
number = {7200},
abstract = {Intermetallic compounds (IMCs) with ordered atomic structure have gained great attention as nanocatalysts for its enhanced activity and stability. Although the reliance of IMC preparation on high-temperature annealing is well known, a comprehensive understanding of the formation mechanisms of IMCs in this process is currently lacking. Here, we employ aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) to track the formation process of IMCs on carbon supports during in-situ annealing, by taking PtFe as a case study within an industry-relevant impregnation synthesis framework. We directly discern five different stages at the atomic level: initial atomic precursors; Pt cluster formation; Pt-Fe disordered alloying; structurally ordered Pt3Fe formation, and final Pt3Fe-PtFe IMC conversion. In particular, we find that the crucial role of high-temperature annealing resides in facilitating the diffusion of Fe towards Pt, enabling the creation of alloys with the targeted stoichiometric ratio, which in turn provides the thermodynamic driving force for the disorder-to-order transition.},
note = {The mechanisms of intermetallic compound (IMC) formation during annealing are poorly understood. Here, the authors identify five stages of PtFe-IMC formation at the atomic level using in-situ STEM, emphasizing the role of high temperature in achieving the targeted stoichiometric ratio.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Intermetallic compounds (IMCs) with ordered atomic structure have gained great attention as nanocatalysts for its enhanced activity and stability. Although the reliance of IMC preparation on high-temperature annealing is well known, a comprehensive understanding of the formation mechanisms of IMCs in this process is currently lacking. Here, we employ aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) to track the formation process of IMCs on carbon supports during in-situ annealing, by taking PtFe as a case study within an industry-relevant impregnation synthesis framework. We directly discern five different stages at the atomic level: initial atomic precursors; Pt cluster formation; Pt-Fe disordered alloying; structurally ordered Pt3Fe formation, and final Pt3Fe-PtFe IMC conversion. In particular, we find that the crucial role of high-temperature annealing resides in facilitating the diffusion of Fe towards Pt, enabling the creation of alloys with the targeted stoichiometric ratio, which in turn provides the thermodynamic driving force for the disorder-to-order transition. |
| 175. | | Xian-Kai Wan, Xu-Shuang Han, Zong-Jie Guan, Wan-Qi Shi, Jiao-Jiao Li, Quan-Ming Wang Interplay of kernel shape and surface structure for NIR luminescence in atomically precise gold nanorods In: Nature Communications, vol. 15, no. 7214, 2024, (Gold nanoclusters with strong emissions in the near-infrared are challenging to attain. Here, the authors show that for rod-shaped nanoclusters, both the shape of the kernel and the rigid surface structure are important to increase the NIR emissions.
). @article{nokey,
title = {Interplay of kernel shape and surface structure for NIR luminescence in atomically precise gold nanorods},
author = {Xian-Kai Wan and Xu-Shuang Han and Zong-Jie Guan and Wan-Qi Shi and Jiao-Jiao Li and Quan-Ming Wang },
url = {https://www.nature.com/articles/s41467-024-51642-w.pdf},
doi = {10.1038/s41467-024-51642-w},
year = {2024},
date = {2024-08-22},
journal = {Nature Communications},
volume = {15},
number = {7214},
abstract = {It is challenging to attain strong near-infrared (NIR) emissive gold nanoclusters. Here we show a rod-shaped cluster with the composition of [Au28(p-MBT)14(Hdppa)3](SO3CF3)2 (1 for short, Hdppa is N,N-bis(diphenylphosphino)amine, p-MBT is 4-methylbenzenethiolate) has been synthesized. Single crystal X-ray structural analysis reveals that it has a rod-like face-centered cubic (fcc) Au22 kernel built from two interpenetrating bicapped cuboctahedral Au15 units. 1 features NIR luminescence with an emission maximum at 920 nm, and the photoluminescence quantum yield (PLQY) is 12%, which is 30-fold of [Au21(m-MBT)12(Hdppa)2]SO3CF3 (2, m-MBT is 3-methylbenzenethiolate) with a similar composition and 60-fold of Au30S(S‑t‑Bu)18 with a similar structure. time-dependent DFT(TDDFT)calculations reveal that the luminescence of 1 is associated with the Au22 kernel. The small Stokes shift of 1 indicates that it has a very small excited state structural distortion, leading to high radiative decay rate (kr) probability. The emission of cluster 1 is a mixture of phosphorescence and thermally activated delayed fluorescence(TADF), and the enhancement of the NIR emission is mainly due to the promotion of kr rather than the inhibition of knr. This work demonstrates that the metal kernel and the surface structure are both very important for cluster-based NIR luminescence materials.},
note = {Gold nanoclusters with strong emissions in the near-infrared are challenging to attain. Here, the authors show that for rod-shaped nanoclusters, both the shape of the kernel and the rigid surface structure are important to increase the NIR emissions.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
It is challenging to attain strong near-infrared (NIR) emissive gold nanoclusters. Here we show a rod-shaped cluster with the composition of [Au28(p-MBT)14(Hdppa)3](SO3CF3)2 (1 for short, Hdppa is N,N-bis(diphenylphosphino)amine, p-MBT is 4-methylbenzenethiolate) has been synthesized. Single crystal X-ray structural analysis reveals that it has a rod-like face-centered cubic (fcc) Au22 kernel built from two interpenetrating bicapped cuboctahedral Au15 units. 1 features NIR luminescence with an emission maximum at 920 nm, and the photoluminescence quantum yield (PLQY) is 12%, which is 30-fold of [Au21(m-MBT)12(Hdppa)2]SO3CF3 (2, m-MBT is 3-methylbenzenethiolate) with a similar composition and 60-fold of Au30S(S‑t‑Bu)18 with a similar structure. time-dependent DFT(TDDFT)calculations reveal that the luminescence of 1 is associated with the Au22 kernel. The small Stokes shift of 1 indicates that it has a very small excited state structural distortion, leading to high radiative decay rate (kr) probability. The emission of cluster 1 is a mixture of phosphorescence and thermally activated delayed fluorescence(TADF), and the enhancement of the NIR emission is mainly due to the promotion of kr rather than the inhibition of knr. This work demonstrates that the metal kernel and the surface structure are both very important for cluster-based NIR luminescence materials. |
| 174. | | Chuan He, Jingzhuo Zhou, Rui Zhou, Cong Chen, Siyi Jing, Kaiyu Mu, Yu-Ting Huang, Chih-Chun Chung, Sheng-Jye Cherng, Yang Lu, King-Ning Tu, Shien-Ping Feng Nanocrystalline copper for direct copper-to-copper bonding with improved cross-interface formation at low thermal budget In: Nature Communications, vol. 15, no. 7095, 2024, (Direct copper-to-copper bonding is crucial for advanced electronic packaging. Here, the authors demonstrate Cu-Cu bonding with low thermal budget using nanocrystalline Cu with a double-layer structure.). @article{nokey,
title = {Nanocrystalline copper for direct copper-to-copper bonding with improved cross-interface formation at low thermal budget},
author = {Chuan He and Jingzhuo Zhou and Rui Zhou and Cong Chen and Siyi Jing and Kaiyu Mu and Yu-Ting Huang and Chih-Chun Chung and Sheng-Jye Cherng and Yang Lu and King-Ning Tu and Shien-Ping Feng },
url = {https://www.nature.com/articles/s41467-024-51510-7.pdf},
doi = {10.1038/s41467-024-51510-7},
year = {2024},
date = {2024-08-17},
journal = {Nature Communications},
volume = {15},
number = {7095},
abstract = {Direct copper-to-copper (Cu-Cu) bonding is a promising technology for advanced electronic packaging. Nanocrystalline (NC) Cu receives increasing attention due to its unique ability to promote grain growth across the bonding interface. However, achieving sufficient grain growth still requires a high thermal budget. This study explores how reducing grain size and controlling impurity concentration in NC Cu leads to substantial grain growth at low temperatures. The fabricated NC Cu has a uniform nanograin size of around 50 nm and a low impurity level of 300 ppm. To prevent ungrown NC and void formation caused by impurity aggregation, we propose a double-layer (DL) structure comprising a normal coarse-grained (CG) layer underneath the NC layer. The CG layer, with a grain size of 1 μm and an impurity level of 3 ppm, acts as a sink, facilitating impurity diffusion from the NC layer to the CG layer. Thanks to sufficient grain growth throughout the entire NC layer, cross-interface Cu-Cu bonding becomes possible under a low thermal budget, either at 100 °C for 60 min or at 200 °C for only 5 min.},
note = {Direct copper-to-copper bonding is crucial for advanced electronic packaging. Here, the authors demonstrate Cu-Cu bonding with low thermal budget using nanocrystalline Cu with a double-layer structure.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Direct copper-to-copper (Cu-Cu) bonding is a promising technology for advanced electronic packaging. Nanocrystalline (NC) Cu receives increasing attention due to its unique ability to promote grain growth across the bonding interface. However, achieving sufficient grain growth still requires a high thermal budget. This study explores how reducing grain size and controlling impurity concentration in NC Cu leads to substantial grain growth at low temperatures. The fabricated NC Cu has a uniform nanograin size of around 50 nm and a low impurity level of 300 ppm. To prevent ungrown NC and void formation caused by impurity aggregation, we propose a double-layer (DL) structure comprising a normal coarse-grained (CG) layer underneath the NC layer. The CG layer, with a grain size of 1 μm and an impurity level of 3 ppm, acts as a sink, facilitating impurity diffusion from the NC layer to the CG layer. Thanks to sufficient grain growth throughout the entire NC layer, cross-interface Cu-Cu bonding becomes possible under a low thermal budget, either at 100 °C for 60 min or at 200 °C for only 5 min. |
| 173. | | Geon Yeong Kim, Shinho Kim, Ki Hyun Park, Hanhwi Jang, Moohyun Kim, Tae Won Nam, Kyeong Min Song, Hongjoo Shin, Yemin Park, Yeongin Cho, Jihyeon Yeom, Min-Jae Choi, Min Seok Jang, Yeon Sik Jung Chiral 3D structures through multi-dimensional transfer printing of multilayer quantum dot patterns In: Nature Communications, vol. 15, no. 6996, 2024, (3D photonic nanostructures can manipulate the amplitude, phase, and polarization of light, but their bottom-up fabrication is hindered by limited structural control. Here, the authors present chiral 3D structures through multi-dimensional transfer printing of multilayer quantum dot patterns.). @article{nokey,
title = {Chiral 3D structures through multi-dimensional transfer printing of multilayer quantum dot patterns},
author = {Geon Yeong Kim and Shinho Kim and Ki Hyun Park and Hanhwi Jang and Moohyun Kim and Tae Won Nam and Kyeong Min Song and Hongjoo Shin and Yemin Park and Yeongin Cho and Jihyeon Yeom and Min-Jae Choi and Min Seok Jang and Yeon Sik Jung },
url = {https://www.nature.com/articles/s41467-024-51179-y.pdf},
doi = {10.1038/s41467-024-51179-y},
year = {2024},
date = {2024-08-14},
journal = {Nature Communications},
volume = {15},
number = {6996},
abstract = {Three-dimensional optical nanostructures have garnered significant interest in photonics due to their extraordinary capabilities to manipulate the amplitude, phase, and polarization states of light. However, achieving complex three-dimensional optical nanostructures with bottom-up fabrication has remained challenging, despite its nanoscale precision and cost-effectiveness, mainly due to inherent limitations in structural controllability. Here, we report the optical characteristics of intricate two- and three-dimensional nanoarchitectures made of colloidal quantum dots fabricated with multi-dimensional transfer printing. Our customizable fabrication platform, directed by tailored interface polarity, enables flexible geometric control over a variety of one-, two-, and three-dimensional quantum dot architectures, achieving tunable and advanced optical features. For example, we demonstrate a two-dimensional quantum dot nanomesh with tuned subwavelength square perforations designed by finite-difference time-domain calculations, achieving an 8-fold enhanced photoluminescence due to the maximized optical resonance. Furthermore, a three-dimensional quantum dot chiral structure is also created via asymmetric stacking of one-dimensional quantum dot layers, realizing a pronounced circular dichroism intensity exceeding 20°.},
note = {3D photonic nanostructures can manipulate the amplitude, phase, and polarization of light, but their bottom-up fabrication is hindered by limited structural control. Here, the authors present chiral 3D structures through multi-dimensional transfer printing of multilayer quantum dot patterns.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Three-dimensional optical nanostructures have garnered significant interest in photonics due to their extraordinary capabilities to manipulate the amplitude, phase, and polarization states of light. However, achieving complex three-dimensional optical nanostructures with bottom-up fabrication has remained challenging, despite its nanoscale precision and cost-effectiveness, mainly due to inherent limitations in structural controllability. Here, we report the optical characteristics of intricate two- and three-dimensional nanoarchitectures made of colloidal quantum dots fabricated with multi-dimensional transfer printing. Our customizable fabrication platform, directed by tailored interface polarity, enables flexible geometric control over a variety of one-, two-, and three-dimensional quantum dot architectures, achieving tunable and advanced optical features. For example, we demonstrate a two-dimensional quantum dot nanomesh with tuned subwavelength square perforations designed by finite-difference time-domain calculations, achieving an 8-fold enhanced photoluminescence due to the maximized optical resonance. Furthermore, a three-dimensional quantum dot chiral structure is also created via asymmetric stacking of one-dimensional quantum dot layers, realizing a pronounced circular dichroism intensity exceeding 20°. |
| 172. | | Yong Zhang, Chenyun He, Qin Yu, Xiao Li, Xiaogang Wang, Yin Zhang, Ji Wang, Chao Jiang, Yunfei Jia, Xian-Cheng Zhang, Binhan Sun, Robert O. Ritchie, Shan-Tung Tu Nacre-like surface nanolaminates enhance fatigue resistance of pure titanium In: Nature Communications, vol. 15, no. 6917, 2024, (Most strategies to improve fatigue resistance address either crack initiation or growth. Here, the authors design a gradient-structured Ti with nacre-like surface nanolaminates that increase fatigue performance by suppressing both stages of cracking). @article{nokey,
title = {Nacre-like surface nanolaminates enhance fatigue resistance of pure titanium},
author = {Yong Zhang and Chenyun He and Qin Yu and Xiao Li and Xiaogang Wang and Yin Zhang and Ji Wang and Chao Jiang and Yunfei Jia and Xian-Cheng Zhang and Binhan Sun and Robert O. Ritchie and Shan-Tung Tu },
url = {https://www.nature.com/articles/s41467-024-51423-5.pdf},
doi = {10.1038/s41467-024-51423-5},
year = {2024},
date = {2024-08-13},
journal = {Nature Communications},
volume = {15},
number = {6917},
abstract = {Fatigue failure is invariably the most crucial failure mode for metallic structural components. Most microstructural strategies for enhancing fatigue resistance are effective in suppressing either crack initiation or propagation, but often do not work for both synergistically. Here, we demonstrate that this challenge can be overcome by architecting a gradient structure featuring a surface layer of nacre-like nanolaminates followed by multi-variant twinned structure in pure titanium. The polarized accommodation of highly regulated grain boundaries in the nanolaminated layer to cyclic loading enhances the structural stability against lamellar thickening and microstructure softening, thereby delaying surface roughening and thus crack nucleation. The decohesion of the nanolaminated grains along horizonal high-angle grain boundaries gives rise to an extraordinarily high frequency (≈1.7 × 103 times per mm) of fatigue crack deflection, effectively reducing fatigue crack propagation rate (by 2 orders of magnitude lower than the homogeneous coarse-grained counterpart). These intriguing features of the surface nanolaminates, along with the various toughening mechanisms activated in the subsurface twinned structure, result in a fatigue resistance that significantly exceeds those of the homogeneous and gradient structures with equiaxed grains. Our work on architecting the surface nanolaminates in gradient structure provides a scalable and sustainable strategy for designing more fatigue-resistant alloys.},
note = {Most strategies to improve fatigue resistance address either crack initiation or growth. Here, the authors design a gradient-structured Ti with nacre-like surface nanolaminates that increase fatigue performance by suppressing both stages of cracking},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fatigue failure is invariably the most crucial failure mode for metallic structural components. Most microstructural strategies for enhancing fatigue resistance are effective in suppressing either crack initiation or propagation, but often do not work for both synergistically. Here, we demonstrate that this challenge can be overcome by architecting a gradient structure featuring a surface layer of nacre-like nanolaminates followed by multi-variant twinned structure in pure titanium. The polarized accommodation of highly regulated grain boundaries in the nanolaminated layer to cyclic loading enhances the structural stability against lamellar thickening and microstructure softening, thereby delaying surface roughening and thus crack nucleation. The decohesion of the nanolaminated grains along horizonal high-angle grain boundaries gives rise to an extraordinarily high frequency (≈1.7 × 103 times per mm) of fatigue crack deflection, effectively reducing fatigue crack propagation rate (by 2 orders of magnitude lower than the homogeneous coarse-grained counterpart). These intriguing features of the surface nanolaminates, along with the various toughening mechanisms activated in the subsurface twinned structure, result in a fatigue resistance that significantly exceeds those of the homogeneous and gradient structures with equiaxed grains. Our work on architecting the surface nanolaminates in gradient structure provides a scalable and sustainable strategy for designing more fatigue-resistant alloys. |
| 171. | | Fenghui Duan, Qian Li, Zhihao Jiang, Lin Zhou, Junhua Luan, Zheling Shen, Weihua Zhou, Shiyuan Zhang, Jie Pan, Xin Zhou, Tao Yang, Jian Lu An order-disorder core-shell strategy for enhanced work-hardening capability and ductility in nanostructured alloys In: Nature Communications, vol. 15, no. 6832, 2024, (Nanocrystalline metallic materials have the merit of high strength, but usually suffer from poor ductility and rapid grain coarsening. Here, the authors develop a nanocrystalline core-shell alloy to overcome these challenges.). @article{nokey,
title = {An order-disorder core-shell strategy for enhanced work-hardening capability and ductility in nanostructured alloys},
author = {Fenghui Duan and Qian Li and Zhihao Jiang and Lin Zhou and Junhua Luan and Zheling Shen and Weihua Zhou and Shiyuan Zhang and Jie Pan and Xin Zhou and Tao Yang and Jian Lu },
url = {https://www.nature.com/articles/s41467-024-50984-9.pdf},
doi = {10.1038/s41467-024-50984-9},
year = {2024},
date = {2024-08-09},
journal = {Nature Communications},
volume = {15},
number = {6832},
abstract = {Nanocrystalline metallic materials have the merit of high strength but usually suffer from poor ductility and rapid grain coarsening, limiting their practical application. Here, we introduce a core-shell nanostructure in a multicomponent alloy to address these challenges simultaneously, achieving a high tensile strength of 2.65 GPa, a large uniform elongation of 17%, and a high thermal stability of 1173 K. Our strategy relies on an ordered superlattice structure that excels in dislocation accumulation, encased by a ≈3 nm disordered face-centered-cubic nanolayer acting as dislocation sources. The ordered superlattice with high anti-phase boundary energy retards dislocation motions, promoting their interaction and storage within the nanograins. The disordered interfacial nanolayer promotes dislocation emission and effectively accommodates the plastic strain at grain boundaries, preventing intergranular cracking. Consequently, the order-disorder core-shell nanostructure exhibits enhanced work-hardening capability and large ductility. Moreover, such core-shell nanostructure exhibits high coarsening resistance at elevated temperatures, enabling it high thermal stability. Such a design strategy holds promise for developing high-performance materials.},
note = {Nanocrystalline metallic materials have the merit of high strength, but usually suffer from poor ductility and rapid grain coarsening. Here, the authors develop a nanocrystalline core-shell alloy to overcome these challenges.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanocrystalline metallic materials have the merit of high strength but usually suffer from poor ductility and rapid grain coarsening, limiting their practical application. Here, we introduce a core-shell nanostructure in a multicomponent alloy to address these challenges simultaneously, achieving a high tensile strength of 2.65 GPa, a large uniform elongation of 17%, and a high thermal stability of 1173 K. Our strategy relies on an ordered superlattice structure that excels in dislocation accumulation, encased by a ≈3 nm disordered face-centered-cubic nanolayer acting as dislocation sources. The ordered superlattice with high anti-phase boundary energy retards dislocation motions, promoting their interaction and storage within the nanograins. The disordered interfacial nanolayer promotes dislocation emission and effectively accommodates the plastic strain at grain boundaries, preventing intergranular cracking. Consequently, the order-disorder core-shell nanostructure exhibits enhanced work-hardening capability and large ductility. Moreover, such core-shell nanostructure exhibits high coarsening resistance at elevated temperatures, enabling it high thermal stability. Such a design strategy holds promise for developing high-performance materials. |
| 170. | | Yihuan Cao, Ming Yang, Qing Du, Fu-Kuo Chiang, Yingjie Zhang, Shi-Wei Chen, Yubin Ke, Hongbo Lou, Fei Zhang, Yuan Wu, Hui Wang, Suihe Jiang, Xiaobin Zhang, Qiaoshi Zeng, Xiongjun Liu, Zhaoping Lu Continuous polyamorphic transition in high-entropy metallic glass In: Nature Communications, no. 6702, 2024, (The understanding of polyamorphic transitions is of scientific and technological importance. Here, the authors report a continuous polyamorphic transition without first-order transition characteristics in high-entropy metallic glasses.). @article{nokey,
title = {Continuous polyamorphic transition in high-entropy metallic glass},
author = {Yihuan Cao and Ming Yang and Qing Du and Fu-Kuo Chiang and Yingjie Zhang and Shi-Wei Chen and Yubin Ke and Hongbo Lou and Fei Zhang and Yuan Wu and Hui Wang and Suihe Jiang and Xiaobin Zhang and Qiaoshi Zeng and Xiongjun Liu and Zhaoping Lu },
url = {https://www.nature.com/articles/s41467-024-51080-8.pdf},
doi = {10.1038/s41467-024-51080-8},
year = {2024},
date = {2024-08-07},
journal = {Nature Communications},
number = {6702},
abstract = {Polyamorphic transition (PT) is a compelling and pivotal physical phenomenon in the field of glass and materials science. Understanding this transition is of scientific and technological significance, as it offers an important pathway for effectively tuning the structure and property of glasses. In contrast to the PT observed in conventional metallic glasses (MGs), which typically exhibit a pronounced first-order nature, herein we report a continuous PT (CPT) without first-order characteristics in high-entropy MGs (HEMGs) upon heating. This CPT behavior is featured by the continuous structural evolution at the atomic level and an increasing chemical concentration gradient with temperature, but no abrupt reduction in volume and energy. The continuous transformation is associated with the absence of local favorable structures and chemical heterogeneity caused by the high configurational entropy, which limits the distance and frequency of atomic diffusion. As a result of the CPT, numerous glass states can be generated, which provides an opportunity to understand the nature, atomic packing, formability, and properties of MGs. Moreover, this discovery highlights the implication of configurational entropy in exploring polyamorphic glasses with an identical composition but highly tunable structures and properties.},
note = {The understanding of polyamorphic transitions is of scientific and technological importance. Here, the authors report a continuous polyamorphic transition without first-order transition characteristics in high-entropy metallic glasses.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Polyamorphic transition (PT) is a compelling and pivotal physical phenomenon in the field of glass and materials science. Understanding this transition is of scientific and technological significance, as it offers an important pathway for effectively tuning the structure and property of glasses. In contrast to the PT observed in conventional metallic glasses (MGs), which typically exhibit a pronounced first-order nature, herein we report a continuous PT (CPT) without first-order characteristics in high-entropy MGs (HEMGs) upon heating. This CPT behavior is featured by the continuous structural evolution at the atomic level and an increasing chemical concentration gradient with temperature, but no abrupt reduction in volume and energy. The continuous transformation is associated with the absence of local favorable structures and chemical heterogeneity caused by the high configurational entropy, which limits the distance and frequency of atomic diffusion. As a result of the CPT, numerous glass states can be generated, which provides an opportunity to understand the nature, atomic packing, formability, and properties of MGs. Moreover, this discovery highlights the implication of configurational entropy in exploring polyamorphic glasses with an identical composition but highly tunable structures and properties. |
| 169. | | Prahlad K. Routh, Evgeniy Redekop, Sebastian Prodinger, Jessi E. S. van der Hoeven, Kang Rui Garrick Lim, Joanna Aizenberg, Maarten Nachtegaal, Adam H. Clark, Anatoly I. Frenkel Restructuring dynamics of surface species in bimetallic nanoparticles probed by modulation excitation spectroscopy In: Nature Communications, vol. 15, no. 6734, 2024, (Nanocatalysts can restructure during reactions. Here, the authors dynamically varied the stoichiometry of the surface species in 30 % Pd-in-Au nanoparticles by modulating H2 and O2 gases and quantified the formation kinetics of Pd regions and oxides.). @article{nokey,
title = {Restructuring dynamics of surface species in bimetallic nanoparticles probed by modulation excitation spectroscopy},
author = {Prahlad K. Routh and Evgeniy Redekop and Sebastian Prodinger and Jessi E. S. van der Hoeven and Kang Rui Garrick Lim and Joanna Aizenberg and Maarten Nachtegaal and Adam H. Clark and Anatoly I. Frenkel },
url = {https://www.nature.com/articles/s41467-024-51068-4.pdf},
doi = {10.1038/s41467-024-51068-4},
year = {2024},
date = {2024-08-07},
journal = {Nature Communications},
volume = {15},
number = {6734},
abstract = {Restructuring of metal components on bimetallic nanoparticle surfaces in response to the changes in reactive environment is a ubiquitous phenomenon whose potential for the design of tunable catalysts is underexplored. The main challenge is the lack of knowledge of the structure, composition, and evolution of species on the nanoparticle surfaces during reaction. We apply a modulation excitation approach to the X-ray absorption spectroscopy of the 30 atomic % Pd in Au supported nanocatalysts via the gas (H2 and O2) concentration modulation. For interpreting restructuring kinetics, we correlate the phase-sensitive detection with the time-domain analysis aided by a denoising algorithm. Here we show that the surface and near-surface species such as Pd oxides and atomically dispersed Pd restructured periodically, featuring different time delays. We propose a model that Pd oxide formation is preceded by the build-up of Pd regions caused by oxygen-driven segregation of Pd atoms towards the surface. During the H2 pulse, rapid reduction and dissolution of Pd follows an induction period which we attribute to H2 dissociation. Periodic perturbations of nanocatalysts by gases can, therefore, enable variations in the stoichiometry of the surface and near-surface oxides and dynamically tune the degree of oxidation/reduction of metals at/near the catalyst surface.},
note = {Nanocatalysts can restructure during reactions. Here, the authors dynamically varied the stoichiometry of the surface species in 30 % Pd-in-Au nanoparticles by modulating H2 and O2 gases and quantified the formation kinetics of Pd regions and oxides.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Restructuring of metal components on bimetallic nanoparticle surfaces in response to the changes in reactive environment is a ubiquitous phenomenon whose potential for the design of tunable catalysts is underexplored. The main challenge is the lack of knowledge of the structure, composition, and evolution of species on the nanoparticle surfaces during reaction. We apply a modulation excitation approach to the X-ray absorption spectroscopy of the 30 atomic % Pd in Au supported nanocatalysts via the gas (H2 and O2) concentration modulation. For interpreting restructuring kinetics, we correlate the phase-sensitive detection with the time-domain analysis aided by a denoising algorithm. Here we show that the surface and near-surface species such as Pd oxides and atomically dispersed Pd restructured periodically, featuring different time delays. We propose a model that Pd oxide formation is preceded by the build-up of Pd regions caused by oxygen-driven segregation of Pd atoms towards the surface. During the H2 pulse, rapid reduction and dissolution of Pd follows an induction period which we attribute to H2 dissociation. Periodic perturbations of nanocatalysts by gases can, therefore, enable variations in the stoichiometry of the surface and near-surface oxides and dynamically tune the degree of oxidation/reduction of metals at/near the catalyst surface. |
| 168. | | Xianglong Lyu, Zhiqiang Zheng, Anitha Shiva, Mertcan Han, Cem Balda Dayan, Mingchao Zhang, Metin Sitti Capillary trapping of various nanomaterials on additively manufactured scaffolds for 3D micro-/nanofabrication In: Nature Communications, vol. 15, no. 6693, 2024, (High-precision 3D micro-/nanofabrication technologies such as two-photon polymerization are limited to photocurable polymers. Here, the authors report a “capillary-trapping” strategy to fabricate various 3D micro-scaffolds composed of different nanomaterials.). @article{nokey,
title = {Capillary trapping of various nanomaterials on additively manufactured scaffolds for 3D micro-/nanofabrication},
author = {Xianglong Lyu and Zhiqiang Zheng and Anitha Shiva and Mertcan Han and Cem Balda Dayan and Mingchao Zhang and Metin Sitti },
url = {https://www.nature.com/articles/s41467-024-51086-2.pdf},
doi = {10.1038/s41467-024-51086-2},
year = {2024},
date = {2024-08-06},
journal = {Nature Communications},
volume = {15},
number = {6693},
abstract = {High-precision additive manufacturing technologies, such as two-photon polymerization, are mainly limited to photo-curable polymers and currently lacks the possibility to produce multimaterial components. Herein, we report a physically bottom-up assembly strategy that leverages capillary force to trap various nanomaterials and assemble them onto three-dimensional (3D) microscaffolds. This capillary-trapping strategy enables precise and uniform assembly of nanomaterials into versatile 3D microstructures with high uniformity and mass loading. Our approach applies to diverse materials irrespective of their physiochemical properties, including polymers, metals, metal oxides, and others. It can integrate at least four different material types into a single 3D microstructure in a sequential, layer-by-layer manner, opening immense possibilities for tailored functionalities on demand. Furthermore, the 3D microscaffolds are removable, facilitating the creation of pure material-based 3D microstructures. This universal 3D micro-/nanofabrication technique with various nanomaterials enables the creation of advanced miniature devices with potential applications in multifunctional microrobots and smart micromachines.},
note = {High-precision 3D micro-/nanofabrication technologies such as two-photon polymerization are limited to photocurable polymers. Here, the authors report a “capillary-trapping” strategy to fabricate various 3D micro-scaffolds composed of different nanomaterials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
High-precision additive manufacturing technologies, such as two-photon polymerization, are mainly limited to photo-curable polymers and currently lacks the possibility to produce multimaterial components. Herein, we report a physically bottom-up assembly strategy that leverages capillary force to trap various nanomaterials and assemble them onto three-dimensional (3D) microscaffolds. This capillary-trapping strategy enables precise and uniform assembly of nanomaterials into versatile 3D microstructures with high uniformity and mass loading. Our approach applies to diverse materials irrespective of their physiochemical properties, including polymers, metals, metal oxides, and others. It can integrate at least four different material types into a single 3D microstructure in a sequential, layer-by-layer manner, opening immense possibilities for tailored functionalities on demand. Furthermore, the 3D microscaffolds are removable, facilitating the creation of pure material-based 3D microstructures. This universal 3D micro-/nanofabrication technique with various nanomaterials enables the creation of advanced miniature devices with potential applications in multifunctional microrobots and smart micromachines. |
| 167. | | Yongqiang Li, Siwei Yang, Wancheng Bao, Quan Tao, Xiuyun Jiang, Jipeng Li, Peng He, Gang Wang, Kai Qi, Hui Dong, Guqiao Ding, Xiaoming Xie Accelerated proton dissociation in an excited state induces superacidic microenvironments around graphene quantum dots In: Nature Communications, vol. 15, no. 6634, 2024, (Understanding interfacial proton transport in an excited state is crucial for catalytic and diagnostic applications of nanomaterials. Here, the authors combine ultra-low-field NMR relaxometry with a light source to study the light-induced proton dissociation of graphene quantum dots.). @article{nokey,
title = {Accelerated proton dissociation in an excited state induces superacidic microenvironments around graphene quantum dots},
author = {Yongqiang Li and Siwei Yang and Wancheng Bao and Quan Tao and Xiuyun Jiang and Jipeng Li and Peng He and Gang Wang and Kai Qi and Hui Dong and Guqiao Ding and Xiaoming Xie },
url = {https://www.nature.com/articles/s41467-024-50982-x.pdf},
doi = {10.1038/s41467-024-50982-x},
year = {2024},
date = {2024-08-05},
journal = {Nature Communications},
volume = {15},
number = {6634},
abstract = {Investigating proton transport at the interface in an excited state facilitates the mechanistic investigation and utilization of nanomaterials. However, there is a lack of suitable tools for in-situ and interfacial analysis. Here we addresses this gap by in-situ observing the proton transport of graphene quantum dots (GQDs) in an excited state through reduction of magnetic resonance relaxation time. Experimental results, utilizing 0.1 mT ultra-low-field nuclear magnetic resonance relaxometry compatible with a light source, reveal the light-induced proton dissociation and acidity of GQDs’ microenvironment in the excited state (Hammett acidity function: –13.40). Theoretical calculations demonstrate significant acidity enhancement in –OH functionalized GQDs with light induction (pKa* = –4.62, stronger than that of H2SO4). Simulations highlight the contributions of edge and phenolic –OH groups to proton dissociation. The light-induced superacidic microenvironment of GQDs benefits functionalization and improves the catalytic performances of GQDs. Importantly, this work advances the understanding of interfacial properties of light-induced sp2–sp3 carbon nanostructure and provides a valuable tool for exploring catalyst interfaces in photocatalysis.},
note = {Understanding interfacial proton transport in an excited state is crucial for catalytic and diagnostic applications of nanomaterials. Here, the authors combine ultra-low-field NMR relaxometry with a light source to study the light-induced proton dissociation of graphene quantum dots.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Investigating proton transport at the interface in an excited state facilitates the mechanistic investigation and utilization of nanomaterials. However, there is a lack of suitable tools for in-situ and interfacial analysis. Here we addresses this gap by in-situ observing the proton transport of graphene quantum dots (GQDs) in an excited state through reduction of magnetic resonance relaxation time. Experimental results, utilizing 0.1 mT ultra-low-field nuclear magnetic resonance relaxometry compatible with a light source, reveal the light-induced proton dissociation and acidity of GQDs’ microenvironment in the excited state (Hammett acidity function: –13.40). Theoretical calculations demonstrate significant acidity enhancement in –OH functionalized GQDs with light induction (pKa* = –4.62, stronger than that of H2SO4). Simulations highlight the contributions of edge and phenolic –OH groups to proton dissociation. The light-induced superacidic microenvironment of GQDs benefits functionalization and improves the catalytic performances of GQDs. Importantly, this work advances the understanding of interfacial properties of light-induced sp2–sp3 carbon nanostructure and provides a valuable tool for exploring catalyst interfaces in photocatalysis. |
| 166. |  | Outhmane Chahib, Yuling Yin, Jung-Ching Liu, Chao Li, Thilo Glatzel, Feng Ding, Qinghong Yuan, Ernst Meyer, Rémy Pawlak Probing charge redistribution at the interface of self-assembled cyclo-P5 pentamers on Ag(111) In: Nature Communications, vol. 15, no. 6542, 2024, (Although unstable in nature, phosphorus pentamers (cyclo-P5) can be synthesized on a silver surface. Here, the authors use scanning probe microscopy to probe charge redistribution at the P5/Ag interface offering potential applications in transistors or solar cells.). @article{nokey,
title = {Probing charge redistribution at the interface of self-assembled cyclo-P5 pentamers on Ag(111)},
author = {Outhmane Chahib and Yuling Yin and Jung-Ching Liu and Chao Li and Thilo Glatzel and Feng Ding and Qinghong Yuan and Ernst Meyer and Rémy Pawlak },
url = {https://www.nature.com/articles/s41467-024-50862-4.pdf},
doi = {10.1038/s41467-024-50862-4},
year = {2024},
date = {2024-08-02},
urldate = {2024-08-02},
journal = {Nature Communications},
volume = {15},
number = {6542},
abstract = {Phosphorus pentamers (cyclo-P5) are unstable in nature but can be synthesized at the Ag(111) surface. Unlike monolayer black phosphorous, little is known about their electronic properties when in contact with metal electrodes, although this is crucial for future applications. Here, we characterize the atomic structure of cyclo-P5 assembled on Ag(111) using atomic force microscopy with functionalized tips and density functional theory. Combining force and tunneling spectroscopy, we find that a strong charge transfer induces an inward dipole moment at the cyclo-P5/Ag interface as well as the formation of an interface state. We probe the image potential states by field-effect resonant tunneling and quantify the increase of the local change of work function of 0.46 eV at the cyclo-P5 assembly. Our experimental approach suggest that the cyclo-P5/Ag interface has the characteristic ingredients of a p-type semiconductor-metal Schottky junction with potential applications in field-effect transistors, diodes, or solar cells.},
note = {Although unstable in nature, phosphorus pentamers (cyclo-P5) can be synthesized on a silver surface. Here, the authors use scanning probe microscopy to probe charge redistribution at the P5/Ag interface offering potential applications in transistors or solar cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Phosphorus pentamers (cyclo-P5) are unstable in nature but can be synthesized at the Ag(111) surface. Unlike monolayer black phosphorous, little is known about their electronic properties when in contact with metal electrodes, although this is crucial for future applications. Here, we characterize the atomic structure of cyclo-P5 assembled on Ag(111) using atomic force microscopy with functionalized tips and density functional theory. Combining force and tunneling spectroscopy, we find that a strong charge transfer induces an inward dipole moment at the cyclo-P5/Ag interface as well as the formation of an interface state. We probe the image potential states by field-effect resonant tunneling and quantify the increase of the local change of work function of 0.46 eV at the cyclo-P5 assembly. Our experimental approach suggest that the cyclo-P5/Ag interface has the characteristic ingredients of a p-type semiconductor-metal Schottky junction with potential applications in field-effect transistors, diodes, or solar cells. |
| 165. |  | Sifan You, Cuiju Yu, Yixuan Gao, Xuechao Li, Guyue Peng, Kaifeng Niu, Jiahao Xi, Chaojie Xu, Shixuan Du, Xingxing Li, Jinlong Yang, Lifeng Chi Quantifying the conductivity of a single polyene chain by lifting with an STM tip In: Nature Communications, vol. 15, no. 6475, 2024, (Polyene is a segment of polyacetylene, a conductive polymer. Here, the authors measured the conductance of single molecular chain of trans-polyene and found a high conductivity and low decay constant, attributed to the alignment of the energy levels.). @article{nokey,
title = {Quantifying the conductivity of a single polyene chain by lifting with an STM tip},
author = {Sifan You and Cuiju Yu and Yixuan Gao and Xuechao Li and Guyue Peng and Kaifeng Niu and Jiahao Xi and Chaojie Xu and Shixuan Du and Xingxing Li and Jinlong Yang and Lifeng Chi },
url = {https://www.nature.com/articles/s41467-024-50915-8.pdf},
doi = {10.1038/s41467-024-50915-8},
year = {2024},
date = {2024-08-01},
urldate = {2024-08-01},
journal = {Nature Communications},
volume = {15},
number = {6475},
abstract = {Conjugated polymers are promising candidates for molecular wires in nanoelectronics, with flexibility in mechanics, stability in chemistry and variety in electrical conductivity. Polyene, as a segment of polyacetylene, is a typical conjugated polymer with straightforward structure and wide-range adjustable conductance. To obtain atomic scale understanding of charge transfer in polyene, we have measured the conductance of a single polyene-based molecular chain via lifting it up with scanning tunneling microscopy tip. Different from semiconducting characters in pristine polyene (polyacetylene), high conductance and low decay constant are obtained, along with an electronic state around Fermi level and characteristic vibrational mode. These observed phenomena result from pinned molecular orbital owing to molecule-electrode coupling at the interface, and weakened bond length alternation due to electron-phonon coupling inside single molecular chain. Our findings emphasize the interfacial characteristics in molecular junctions and promising properties of polyene, with single molecular conductance as a vital tool for bringing insights into the design and construction of nanodevices.},
note = {Polyene is a segment of polyacetylene, a conductive polymer. Here, the authors measured the conductance of single molecular chain of trans-polyene and found a high conductivity and low decay constant, attributed to the alignment of the energy levels.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Conjugated polymers are promising candidates for molecular wires in nanoelectronics, with flexibility in mechanics, stability in chemistry and variety in electrical conductivity. Polyene, as a segment of polyacetylene, is a typical conjugated polymer with straightforward structure and wide-range adjustable conductance. To obtain atomic scale understanding of charge transfer in polyene, we have measured the conductance of a single polyene-based molecular chain via lifting it up with scanning tunneling microscopy tip. Different from semiconducting characters in pristine polyene (polyacetylene), high conductance and low decay constant are obtained, along with an electronic state around Fermi level and characteristic vibrational mode. These observed phenomena result from pinned molecular orbital owing to molecule-electrode coupling at the interface, and weakened bond length alternation due to electron-phonon coupling inside single molecular chain. Our findings emphasize the interfacial characteristics in molecular junctions and promising properties of polyene, with single molecular conductance as a vital tool for bringing insights into the design and construction of nanodevices. |
| 164. | 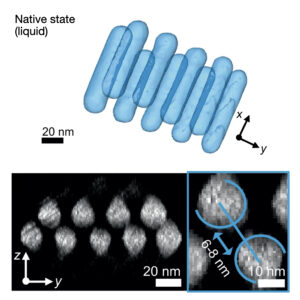 | Daniel Arenas Esteban, Da Wang, Ajinkya Kadu, Noa Olluyn, Ana Sánchez-Iglesias, Alejandro Gomez-Perez, Jesús González-Casablanca, Stavros Nicolopoulos, Luis M. Liz-Marzán, Sara Bals Quantitative 3D structural analysis of small colloidal assemblies under native conditions by liquid-cell fast electron tomography In: Nature Communications, vol. 15, no. 6399, 2024, (Drying force-induced deformation complicates the characterization of the 3D structure of colloidal assemblies. Here, the authors develop a liquid electron tomography method for unravelling the 3D structures of small colloidal assemblies under native conditions.). @article{nokey,
title = {Quantitative 3D structural analysis of small colloidal assemblies under native conditions by liquid-cell fast electron tomography},
author = {Daniel Arenas Esteban and Da Wang and Ajinkya Kadu and Noa Olluyn and Ana Sánchez-Iglesias and Alejandro Gomez-Perez and Jesús González-Casablanca and Stavros Nicolopoulos and Luis M. Liz-Marzán and Sara Bals },
url = {https://www.nature.com/articles/s41467-024-50652-y.pdf},
doi = {10.1038/s41467-024-50652-y},
year = {2024},
date = {2024-07-30},
urldate = {2024-07-30},
journal = {Nature Communications},
volume = {15},
number = {6399},
abstract = {Electron tomography has become a commonly used tool to investigate the three-dimensional (3D) structure of nanomaterials, including colloidal nanoparticle assemblies. However, electron microscopy is typically done under high-vacuum conditions, requiring sample preparation for assemblies obtained by wet colloid chemistry methods. This involves solvent evaporation and deposition on a solid support, which consistently alters the nanoparticle organization. Here, we suggest using electron tomography to study nanoparticle assemblies in their original colloidal liquid environment. To address the challenges related to electron tomography in liquid, we devise a method that combines fast data acquisition in a commercial liquid-cell with a dedicated alignment and reconstruction workflow. We present the advantages of this methodology in accurately characterizing two different systems. 3D reconstructions of assemblies comprising polystyrene-capped Au nanoparticles encapsulated in polymeric shells reveal less compact and more distorted configurations for experiments performed in a liquid medium compared to their dried counterparts. A similar expansion can be observed in quantitative analysis of the surface-to-surface distances of self-assembled Au nanorods in water rather than in a vacuum, in agreement with bulk measurements. This study, therefore, emphasizes the importance of developing high-resolution characterization tools that preserve the native environment of colloidal nanostructures.},
note = {Drying force-induced deformation complicates the characterization of the 3D structure of colloidal assemblies. Here, the authors develop a liquid electron tomography method for unravelling the 3D structures of small colloidal assemblies under native conditions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Electron tomography has become a commonly used tool to investigate the three-dimensional (3D) structure of nanomaterials, including colloidal nanoparticle assemblies. However, electron microscopy is typically done under high-vacuum conditions, requiring sample preparation for assemblies obtained by wet colloid chemistry methods. This involves solvent evaporation and deposition on a solid support, which consistently alters the nanoparticle organization. Here, we suggest using electron tomography to study nanoparticle assemblies in their original colloidal liquid environment. To address the challenges related to electron tomography in liquid, we devise a method that combines fast data acquisition in a commercial liquid-cell with a dedicated alignment and reconstruction workflow. We present the advantages of this methodology in accurately characterizing two different systems. 3D reconstructions of assemblies comprising polystyrene-capped Au nanoparticles encapsulated in polymeric shells reveal less compact and more distorted configurations for experiments performed in a liquid medium compared to their dried counterparts. A similar expansion can be observed in quantitative analysis of the surface-to-surface distances of self-assembled Au nanorods in water rather than in a vacuum, in agreement with bulk measurements. This study, therefore, emphasizes the importance of developing high-resolution characterization tools that preserve the native environment of colloidal nanostructures. |
| 163. | 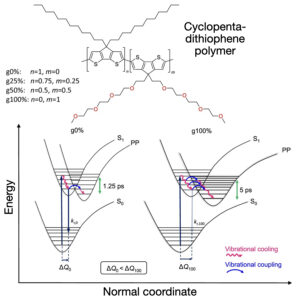 | Katia Pagano, Jin Gwan Kim, Joel Luke, Ellasia Tan, Katherine Stewart, Igor V. Sazanovich, Gabriel Karras, Hristo Ivov Gonev, Adam V. Marsh, Na Yeong Kim, Sooncheol Kwon, Young Yong Kim, M. Isabel Alonso, Bernhard Dörling, Mariano Campoy-Quiles, Anthony W. Parker, Tracey M. Clarke, Yun-Hi Kim, Ji-Seon Kim Slow vibrational relaxation drives ultrafast formation of photoexcited polaron pair states in glycolated conjugated polymers In: Nature Communications, vol. 15, no. 6153, 2024, (Glycol sidechains are often used to enhance the performance of organic photoconversion and electrochemical devices. Here, the authors provide photophysical insight into the role of glycol sidechains for the formation of polaron pairs induced by strong vibrational coupling.). @article{nokey,
title = {Slow vibrational relaxation drives ultrafast formation of photoexcited polaron pair states in glycolated conjugated polymers},
author = {Katia Pagano and Jin Gwan Kim and Joel Luke and Ellasia Tan and Katherine Stewart and Igor V. Sazanovich and Gabriel Karras and Hristo Ivov Gonev and Adam V. Marsh and Na Yeong Kim and Sooncheol Kwon and Young Yong Kim and M. Isabel Alonso and Bernhard Dörling and Mariano Campoy-Quiles and Anthony W. Parker and Tracey M. Clarke and Yun-Hi Kim and Ji-Seon Kim },
url = {https://www.nature.com/articles/s41467-024-50530-7.pdf},
doi = {10.1038/s41467-024-50530-7},
year = {2024},
date = {2024-07-22},
urldate = {2024-07-22},
journal = {Nature Communications},
volume = {15},
number = {6153},
abstract = {Glycol sidechains are often used to enhance the performance of organic photoconversion and electrochemical devices. Herein, we study their effects on electronic states and electronic properties. We find that polymer glycolation not only induces more disordered packing, but also results in a higher reorganisation energy due to more localised π-electron density. Transient absorption spectroscopy and femtosecond stimulated Raman spectroscopy are utilised to monitor the structural relaxation dynamics coupled to the excited state formation upon photoexcitation. Singlet excitons are initially formed, followed by polaron pair formation. The associated structural relaxation slows down in glycolated polymers (5 ps vs. 1.25 ps for alkylated), consistent with larger reorganisation energy. This slower vibrational relaxation is found to drive ultrafast formation of the polaron pair state (5 ps vs. 10 ps for alkylated). These results provide key experimental evidence demonstrating the impact of molecular structure on electronic state formation driven by strong vibrational coupling.},
note = {Glycol sidechains are often used to enhance the performance of organic photoconversion and electrochemical devices. Here, the authors provide photophysical insight into the role of glycol sidechains for the formation of polaron pairs induced by strong vibrational coupling.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Glycol sidechains are often used to enhance the performance of organic photoconversion and electrochemical devices. Herein, we study their effects on electronic states and electronic properties. We find that polymer glycolation not only induces more disordered packing, but also results in a higher reorganisation energy due to more localised π-electron density. Transient absorption spectroscopy and femtosecond stimulated Raman spectroscopy are utilised to monitor the structural relaxation dynamics coupled to the excited state formation upon photoexcitation. Singlet excitons are initially formed, followed by polaron pair formation. The associated structural relaxation slows down in glycolated polymers (5 ps vs. 1.25 ps for alkylated), consistent with larger reorganisation energy. This slower vibrational relaxation is found to drive ultrafast formation of the polaron pair state (5 ps vs. 10 ps for alkylated). These results provide key experimental evidence demonstrating the impact of molecular structure on electronic state formation driven by strong vibrational coupling. |
| 162. | 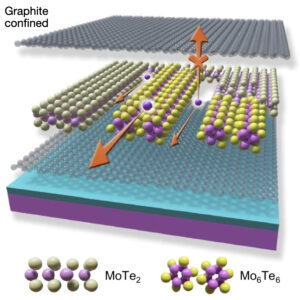 | Qishuo Yang, Yun-Peng Wang, Xiao-Lei Shi, XingXing Li, Erding Zhao, Zhi-Gang Chen, Jin Zou, Kai Leng, Yongqing Cai, Liang Zhu, Sokrates T. Pantelides, Junhao Lin Constrained patterning of orientated metal chalcogenide nanowires and their growth mechanism In: Nature Communications, vol. 15, no. 6074, 2024, (Nanowires made of transition metal chalcogenides are promising as connecting elements in devices, but so far only isotropic growth is feasible. Here, the authors regulate the growth orientation of nanowires by introducing external stimuli into graphite-confined MoTe2 heterostructures.
). @article{nokey,
title = {Constrained patterning of orientated metal chalcogenide nanowires and their growth mechanism},
author = {Qishuo Yang and Yun-Peng Wang and Xiao-Lei Shi and XingXing Li and Erding Zhao and Zhi-Gang Chen and Jin Zou and Kai Leng and Yongqing Cai and Liang Zhu and Sokrates T. Pantelides and Junhao Lin },
url = {https://www.nature.com/articles/s41467-024-50525-4.pdf},
doi = {10.1038/s41467-024-50525-4},
year = {2024},
date = {2024-07-18},
urldate = {2024-07-18},
journal = {Nature Communications},
volume = {15},
number = {6074},
abstract = {One-dimensional metallic transition-metal chalcogenide nanowires (TMC-NWs) hold promise for interconnecting devices built on two-dimensional (2D) transition-metal dichalcogenides, but only isotropic growth has so far been demonstrated. Here we show the direct patterning of highly oriented Mo6Te6 NWs in 2D molybdenum ditelluride (MoTe2) using graphite as confined encapsulation layers under external stimuli. The atomic structural transition is studied through in-situ electrical biasing the fabricated heterostructure in a scanning transmission electron microscope. Atomic resolution high-angle annular dark-field STEM images reveal that the conversion of Mo6Te6 NWs from MoTe2 occurs only along specific directions. Combined with first-principles calculations, we attribute the oriented growth to the local Joule-heating induced by electrical bias near the interface of the graphite-MoTe2 heterostructure and the confinement effect generated by graphite. Using the same strategy, we fabricate oriented NWs confined in graphite as lateral contact electrodes in the 2H-MoTe2 FET, achieving a low Schottky barrier of 11.5 meV, and low contact resistance of 43.7 Ω µm at the metal-NW interface. Our work introduces possible approaches to fabricate oriented NWs for interconnections in flexible 2D nanoelectronics through direct metal phase patterning.},
note = {Nanowires made of transition metal chalcogenides are promising as connecting elements in devices, but so far only isotropic growth is feasible. Here, the authors regulate the growth orientation of nanowires by introducing external stimuli into graphite-confined MoTe2 heterostructures.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
One-dimensional metallic transition-metal chalcogenide nanowires (TMC-NWs) hold promise for interconnecting devices built on two-dimensional (2D) transition-metal dichalcogenides, but only isotropic growth has so far been demonstrated. Here we show the direct patterning of highly oriented Mo6Te6 NWs in 2D molybdenum ditelluride (MoTe2) using graphite as confined encapsulation layers under external stimuli. The atomic structural transition is studied through in-situ electrical biasing the fabricated heterostructure in a scanning transmission electron microscope. Atomic resolution high-angle annular dark-field STEM images reveal that the conversion of Mo6Te6 NWs from MoTe2 occurs only along specific directions. Combined with first-principles calculations, we attribute the oriented growth to the local Joule-heating induced by electrical bias near the interface of the graphite-MoTe2 heterostructure and the confinement effect generated by graphite. Using the same strategy, we fabricate oriented NWs confined in graphite as lateral contact electrodes in the 2H-MoTe2 FET, achieving a low Schottky barrier of 11.5 meV, and low contact resistance of 43.7 Ω µm at the metal-NW interface. Our work introduces possible approaches to fabricate oriented NWs for interconnections in flexible 2D nanoelectronics through direct metal phase patterning. |
| 161. | 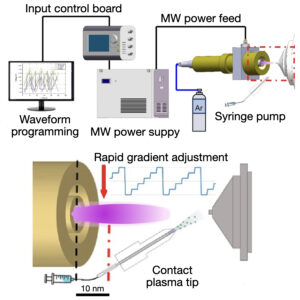 | Fengjian Chu, Gaosheng Zhao, Wei Wei, Nazifi Sani Shuaibu, Hongru Feng, Yuanjiang Pan, Xiaozhi Wang Wide-energy programmable microwave plasma-ionization for high-coverage mass spectrometry analysis In: Nature Communications, vol. 15, no. 6075, 2024, (Ion source technology in mass spectrometry still has limitations in analyte coverage. Here, the authors present a wide-energy programmable microwave plasma ionization mass spectrometry system that enables MS analysis with high coverage.). @article{nokey,
title = {Wide-energy programmable microwave plasma-ionization for high-coverage mass spectrometry analysis},
author = {Fengjian Chu and Gaosheng Zhao and Wei Wei and Nazifi Sani Shuaibu and Hongru Feng and Yuanjiang Pan and Xiaozhi Wang },
url = {https://www.nature.com/articles/s41467-024-50322-z.pdf},
doi = {10.1038/s41467-024-50322-z},
year = {2024},
date = {2024-07-18},
urldate = {2024-07-18},
journal = {Nature Communications},
volume = {15},
number = {6075},
abstract = {Although numerous ambient ionization mass spectroscopy technologies have been developed over the past 20 years to address diverse analytical circumstances, a single-ion source technique that can handle all analyte types is still lacking. Here, a wide-energy programmable microwave plasma-ionization mass spectrometry (WPMPI-MS) system is presented, through which MS analysis can achieve high coverage of substances with various characteristics by digitally regulating the microwave energy. In addition, ionization energy can be rapidly scanned using programmable waveforms, enabling the simultaneous detection of biomolecules, heavy metals, non-polar molecules, etc., in seconds. WPMPI-MS performs well in analyzing real samples, rapidly analyzing nine toxicological standards in one drop of serum, and demonstrating good quantification and liquid chromatography coupling capability. The WPMPI-MS has also been used to detect soil extracts, solid pharmaceuticals, and landfill leachate, further demonstrating its robust analytical capabilities for real samples. The prospective uses of the technology in biological and chemical analysis are extensive, and it is anticipated to emerge as a viable alternative to commercially available ion sources.},
note = {Ion source technology in mass spectrometry still has limitations in analyte coverage. Here, the authors present a wide-energy programmable microwave plasma ionization mass spectrometry system that enables MS analysis with high coverage.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Although numerous ambient ionization mass spectroscopy technologies have been developed over the past 20 years to address diverse analytical circumstances, a single-ion source technique that can handle all analyte types is still lacking. Here, a wide-energy programmable microwave plasma-ionization mass spectrometry (WPMPI-MS) system is presented, through which MS analysis can achieve high coverage of substances with various characteristics by digitally regulating the microwave energy. In addition, ionization energy can be rapidly scanned using programmable waveforms, enabling the simultaneous detection of biomolecules, heavy metals, non-polar molecules, etc., in seconds. WPMPI-MS performs well in analyzing real samples, rapidly analyzing nine toxicological standards in one drop of serum, and demonstrating good quantification and liquid chromatography coupling capability. The WPMPI-MS has also been used to detect soil extracts, solid pharmaceuticals, and landfill leachate, further demonstrating its robust analytical capabilities for real samples. The prospective uses of the technology in biological and chemical analysis are extensive, and it is anticipated to emerge as a viable alternative to commercially available ion sources. |
| 160. | 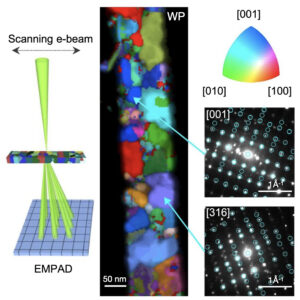 | Gangtae Jin, Christian D. Multunas, James L. Hart, Mehrdad T. Kiani, Nghiep Khoan Duong, Quynh P. Sam, Han Wang, Yeryun Cheon, David J. Hynek, Hyeuk Jin Han, Ravishankar Sundararaman, Judy J. Cha Diameter-dependent phase selectivity in 1D-confined tungsten phosphides In: Nature Communications, vol. 15, no. 5889, 2024, (Topological materials confined in 1D could transform computing technology, but their crystallization is poorly understood. Here, the authors demonstrate template-based synthesis of 1D nanowires, revealing diameter-dependent phase selectivity.). @article{nokey,
title = {Diameter-dependent phase selectivity in 1D-confined tungsten phosphides},
author = {Gangtae Jin and Christian D. Multunas and James L. Hart and Mehrdad T. Kiani and Nghiep Khoan Duong and Quynh P. Sam and Han Wang and Yeryun Cheon and David J. Hynek and Hyeuk Jin Han and Ravishankar Sundararaman and Judy J. Cha },
url = {https://www.nature.com/articles/s41467-024-50323-y.pdf},
doi = {10.1038/s41467-024-50323-y},
year = {2024},
date = {2024-07-13},
urldate = {2024-07-13},
journal = {Nature Communications},
volume = {15},
number = {5889},
abstract = {Topological materials confined in 1D can transform computing technologies, such as 1D topological semimetals for nanoscale interconnects and 1D topological superconductors for fault-tolerant quantum computing. As such, understanding crystallization of 1D-confined topological materials is critical. Here, we demonstrate 1D template-assisted nanowire synthesis where we observe diameter-dependent phase selectivity for tungsten phosphides. A phase bifurcation occurs to produce tungsten monophosphide and tungsten diphosphide at the cross-over nanowire diameter regime of 35–70 nm. Four-dimensional scanning transmission electron microscopy is used to identify the two phases and to map crystallographic orientations of grains at a few nm resolution. The 1D-confined phase selectivity is attributed to the minimization of the total surface energy, which depends on the nanowire diameter and chemical potentials of precursors. Theoretical calculations are carried out to construct the diameter-dependent phase diagram, which agrees with experimental observations. Our findings suggest a crystallization route to stabilize topological materials confined in 1D},
note = {Topological materials confined in 1D could transform computing technology, but their crystallization is poorly understood. Here, the authors demonstrate template-based synthesis of 1D nanowires, revealing diameter-dependent phase selectivity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Topological materials confined in 1D can transform computing technologies, such as 1D topological semimetals for nanoscale interconnects and 1D topological superconductors for fault-tolerant quantum computing. As such, understanding crystallization of 1D-confined topological materials is critical. Here, we demonstrate 1D template-assisted nanowire synthesis where we observe diameter-dependent phase selectivity for tungsten phosphides. A phase bifurcation occurs to produce tungsten monophosphide and tungsten diphosphide at the cross-over nanowire diameter regime of 35–70 nm. Four-dimensional scanning transmission electron microscopy is used to identify the two phases and to map crystallographic orientations of grains at a few nm resolution. The 1D-confined phase selectivity is attributed to the minimization of the total surface energy, which depends on the nanowire diameter and chemical potentials of precursors. Theoretical calculations are carried out to construct the diameter-dependent phase diagram, which agrees with experimental observations. Our findings suggest a crystallization route to stabilize topological materials confined in 1D |
| 159. | 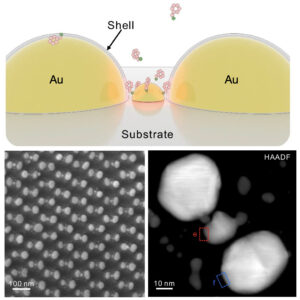 | Xing Zhao, Xiaojing Liu, Dexiang Chen, Guodong Shi, Guoqun Li, Xiao Tang, Xiangnan Zhu, Mingze Li, Lei Yao, Yunjia Wei, Wenzhe Song, Zixuan Sun, Xingce Fan, Zhixin Zhou, Teng Qiu, Qi Hao Plasmonic trimers designed as SERS-active chemical traps for subtyping of lung tumors In: Nature Communications, vol. 15, no. 5855, 2024, (SERS spectroscopy critically relies on the accumulation of analyte molecules in electromagnetic ‘hotspots’. Here, the authors demonstrate plasmonic trimers with central particles that act as chemical traps, enabling the subtyping of lung tumors using fresh tissue samples.). @article{nokey,
title = {Plasmonic trimers designed as SERS-active chemical traps for subtyping of lung tumors},
author = {Xing Zhao and Xiaojing Liu and Dexiang Chen and Guodong Shi and Guoqun Li and Xiao Tang and Xiangnan Zhu and Mingze Li and Lei Yao and Yunjia Wei and Wenzhe Song and Zixuan Sun and Xingce Fan and Zhixin Zhou and Teng Qiu and Qi Hao },
url = {https://www.nature.com/articles/s41467-024-50321-0.pdf},
doi = {10.1038/s41467-024-50321-0},
year = {2024},
date = {2024-07-12},
urldate = {2024-07-12},
journal = {Nature Communications},
volume = {15},
number = {5855},
abstract = {Plasmonic materials can generate strong electromagnetic fields to boost the Raman scattering of surrounding molecules, known as surface-enhanced Raman scattering. However, these electromagnetic fields are heterogeneous, with only molecules located at the ‘hotspots’, which account for ≈ 1% of the surface area, experiencing efficient enhancement. Herein, we propose patterned plasmonic trimers, consisting of a pair of plasmonic dimers at the bilateral sides and a trap particle positioned in between, to address this challenge. The trimer configuration selectively directs probe molecules to the central traps where ‘hotspots’ are located through chemical affinity, ensuring a precise spatial overlap between the probes and the location of maximum field enhancement. We investigate the Raman enhancement of the Au@Al2O3-Au-Au@Al2O3 trimers, achieving a detection limit of 10−14 M of 4-methylbenzenethiol, 4-mercaptopyridine, and 4-aminothiophenol. Moreover, single-molecule SERS sensitivity is demonstrated by a bi-analyte method. Benefiting from this sensitivity, our approach is employed for the early detection of lung tumors using fresh tissues. Our findings suggest that this approach is sensitive to adenocarcinoma but not to squamous carcinoma or benign cases, offering insights into the differentiation between lung tumor subtypes.},
note = {SERS spectroscopy critically relies on the accumulation of analyte molecules in electromagnetic ‘hotspots’. Here, the authors demonstrate plasmonic trimers with central particles that act as chemical traps, enabling the subtyping of lung tumors using fresh tissue samples.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Plasmonic materials can generate strong electromagnetic fields to boost the Raman scattering of surrounding molecules, known as surface-enhanced Raman scattering. However, these electromagnetic fields are heterogeneous, with only molecules located at the ‘hotspots’, which account for ≈ 1% of the surface area, experiencing efficient enhancement. Herein, we propose patterned plasmonic trimers, consisting of a pair of plasmonic dimers at the bilateral sides and a trap particle positioned in between, to address this challenge. The trimer configuration selectively directs probe molecules to the central traps where ‘hotspots’ are located through chemical affinity, ensuring a precise spatial overlap between the probes and the location of maximum field enhancement. We investigate the Raman enhancement of the Au@Al2O3-Au-Au@Al2O3 trimers, achieving a detection limit of 10−14 M of 4-methylbenzenethiol, 4-mercaptopyridine, and 4-aminothiophenol. Moreover, single-molecule SERS sensitivity is demonstrated by a bi-analyte method. Benefiting from this sensitivity, our approach is employed for the early detection of lung tumors using fresh tissues. Our findings suggest that this approach is sensitive to adenocarcinoma but not to squamous carcinoma or benign cases, offering insights into the differentiation between lung tumor subtypes. |
| 158. |  | Sung Hyun Park, Sukyoung Kim, Jae Whan Park, Seunghee Kim, Wonsuk Cha, Joonseok Lee In-situ and wavelength-dependent photocatalytic strain evolution of a single Au nanoparticle on a TiO2 film In: Nature Communications, vol. 15, no. 5416, 2024, (The wavelength-dependent structural deformations of nanoparticles during photocatalysis are poorly understood. Here, the authors present the photocatalytic strain evolution of a single Au nanoparticle using 3D Bragg coherent X-ray diffraction imaging.
). @article{nokey,
title = {In-situ and wavelength-dependent photocatalytic strain evolution of a single Au nanoparticle on a TiO2 film},
author = {Sung Hyun Park and Sukyoung Kim and Jae Whan Park and Seunghee Kim and Wonsuk Cha and Joonseok Lee },
url = {https://www.nature.com/articles/s41467-024-49862-1.pdf},
doi = {10.1038/s41467-024-49862-1},
year = {2024},
date = {2024-06-27},
urldate = {2024-06-27},
journal = {Nature Communications},
volume = {15},
number = {5416},
abstract = {Photocatalysis is a promising technique due to its capacity to efficiently harvest solar energy and its potential to address the global energy crisis. However, the structure–activity relationships of photocatalyst during wavelength-dependent photocatalytic reactions remains largely unexplored because it is difficult to measure under operating conditions. Here we show the photocatalytic strain evolution of a single Au nanoparticle (AuNP) supported on a TiO2 film by combining three-dimensional (3D) Bragg coherent X-ray diffraction imaging with an external light source. The wavelength-dependent generation of reactive oxygen species (ROS) has significant effects on the structural deformation of the AuNP, leading to its strain evolution. Density functional theory (DFT) calculations are employed to rationalize the induced strain caused by the adsorption of ROS on the AuNP surface. These observations provide insights of how the photocatalytic activity impacts on the structural deformation of AuNP, contributing to the general understanding of the atomic-level catalytic adsorption process.},
note = {The wavelength-dependent structural deformations of nanoparticles during photocatalysis are poorly understood. Here, the authors present the photocatalytic strain evolution of a single Au nanoparticle using 3D Bragg coherent X-ray diffraction imaging.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Photocatalysis is a promising technique due to its capacity to efficiently harvest solar energy and its potential to address the global energy crisis. However, the structure–activity relationships of photocatalyst during wavelength-dependent photocatalytic reactions remains largely unexplored because it is difficult to measure under operating conditions. Here we show the photocatalytic strain evolution of a single Au nanoparticle (AuNP) supported on a TiO2 film by combining three-dimensional (3D) Bragg coherent X-ray diffraction imaging with an external light source. The wavelength-dependent generation of reactive oxygen species (ROS) has significant effects on the structural deformation of the AuNP, leading to its strain evolution. Density functional theory (DFT) calculations are employed to rationalize the induced strain caused by the adsorption of ROS on the AuNP surface. These observations provide insights of how the photocatalytic activity impacts on the structural deformation of AuNP, contributing to the general understanding of the atomic-level catalytic adsorption process. |
| 157. |  | Amna Abdeljaoued, Beatriz López Ruiz, Yikalo-Eyob Tecle, Marie Langner, Natalie Bonakdar, Gudrun Bleyer, Patrik Stenner, Nicolas Vogel Efficient removal of nanoplastics from industrial wastewater through synergetic electrophoretic deposition and particle-stabilized foam formation In: Nature Communications, vol. 15, no. 5437, 2024, (Nanoplastics represent a significant environmental challenge due to their minute size, which complicates removal efforts. Here, the authors present a method to effectively extract colloidally stable nanoplastic particles from industrial wastewater.). @article{nokey,
title = {Efficient removal of nanoplastics from industrial wastewater through synergetic electrophoretic deposition and particle-stabilized foam formation},
author = {Amna Abdeljaoued and Beatriz López Ruiz and Yikalo-Eyob Tecle and Marie Langner and Natalie Bonakdar and Gudrun Bleyer and Patrik Stenner and Nicolas Vogel },
url = {https://www.nature.com/articles/s41467-024-48142-2.pdf},
doi = {10.1038/s41467-024-48142-2},
year = {2024},
date = {2024-06-27},
urldate = {2024-06-27},
journal = {Nature Communications},
volume = {15},
number = {5437},
abstract = {Microplastic particles have been discovered in virtually all ecosystems worldwide, yet they may only represent the surface of a much larger issue. Nanoplastics, with dimensions well below 1 µm, pose an even greater environmental concern. Due to their size, they can infiltrate and disrupt individual cells within organisms, potentially exacerbating ecological impacts. Moreover, their minute dimensions present several hurdles for removal, setting them apart from microplastics. Here, we describe a process to remove colloidally stable nanoplastics from wastewater, which synergistically combines electrophoretic deposition and the formation of particle-stabilized foam. This approach capitalizes on localized changes in particle hydrophilicity induced by pH fluctuations resulting from water electrolysis at the electrode surface. By leveraging these pH shifts to enhance particle attachment to nascent bubbles proximal to the electrode, separation of colloidal particles from aqueous dispersions is achieved. Using poly(methyl methacrylate) (PMMA) colloidal particles as a model, we gain insights into the separation mechanisms, which are subsequently applied to alternative model systems with varying surface properties and materials, as well as to real-world industrial wastewaters from dispersion paints and PMMA fabrication processes. Our investigations demonstrate removal efficiencies surpassing 90%.},
note = {Nanoplastics represent a significant environmental challenge due to their minute size, which complicates removal efforts. Here, the authors present a method to effectively extract colloidally stable nanoplastic particles from industrial wastewater.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Microplastic particles have been discovered in virtually all ecosystems worldwide, yet they may only represent the surface of a much larger issue. Nanoplastics, with dimensions well below 1 µm, pose an even greater environmental concern. Due to their size, they can infiltrate and disrupt individual cells within organisms, potentially exacerbating ecological impacts. Moreover, their minute dimensions present several hurdles for removal, setting them apart from microplastics. Here, we describe a process to remove colloidally stable nanoplastics from wastewater, which synergistically combines electrophoretic deposition and the formation of particle-stabilized foam. This approach capitalizes on localized changes in particle hydrophilicity induced by pH fluctuations resulting from water electrolysis at the electrode surface. By leveraging these pH shifts to enhance particle attachment to nascent bubbles proximal to the electrode, separation of colloidal particles from aqueous dispersions is achieved. Using poly(methyl methacrylate) (PMMA) colloidal particles as a model, we gain insights into the separation mechanisms, which are subsequently applied to alternative model systems with varying surface properties and materials, as well as to real-world industrial wastewaters from dispersion paints and PMMA fabrication processes. Our investigations demonstrate removal efficiencies surpassing 90%. |
| 156. | | Hao Li, Tian Wang, Jiaojiao Han, Ying Xu, Xi Kang, Xiaosong Li, Manzhou Zhu Fluorescence resonance energy transfer in atomically precise metal nanoclusters by cocrystallization-induced spatial confinement In: Nature Communications, vol. 15, no. 5351, 2024, (Understanding FRET of metal nanoparticles at the atomic level has long been a challenge. Here, the authors have achieved FRET activity with atomically precise Cu clusters by using a cocrystallisation-induced spatial confinement strategy.). @article{nokey,
title = {Fluorescence resonance energy transfer in atomically precise metal nanoclusters by cocrystallization-induced spatial confinement},
author = {Hao Li and Tian Wang and Jiaojiao Han and Ying Xu and Xi Kang and Xiaosong Li and Manzhou Zhu },
url = {https://www.nature.com/articles/s41467-024-49735-7.pdf},
doi = {10.1038/s41467-024-49735-7},
year = {2024},
date = {2024-06-24},
journal = {Nature Communications},
volume = {15},
number = {5351},
abstract = {Understanding the fluorescence resonance energy transfer (FRET) of metal nanoparticles at the atomic level has long been a challenge due to the lack of accurate systems with definite distance and orientation of molecules. Here we present the realization of achieving FRET between two atomically precise copper nanoclusters through cocrystallization-induced spatial confinement. In this study, we demonstrate the establishment of FRET in a cocrystallized Cu8(p-MBT)8(PPh3)4@Cu10(p-MBT)10(PPh3)4 system by exploiting the overlapping spectra between the excitation of the Cu10(p-MBT)10(PPh3)4 cluster and the emission of the Cu8(p-MBT)8(PPh3)4 cluster, combined with accurate control over the confined space between the two nanoclusters. Density functional theory is employed to provide deeper insights into the role of the distance and dipole orientations of molecules to illustrate the FRET procedure between two cluster molecules at the electronic structure level.},
note = {Understanding FRET of metal nanoparticles at the atomic level has long been a challenge. Here, the authors have achieved FRET activity with atomically precise Cu clusters by using a cocrystallisation-induced spatial confinement strategy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Understanding the fluorescence resonance energy transfer (FRET) of metal nanoparticles at the atomic level has long been a challenge due to the lack of accurate systems with definite distance and orientation of molecules. Here we present the realization of achieving FRET between two atomically precise copper nanoclusters through cocrystallization-induced spatial confinement. In this study, we demonstrate the establishment of FRET in a cocrystallized Cu8(p-MBT)8(PPh3)4@Cu10(p-MBT)10(PPh3)4 system by exploiting the overlapping spectra between the excitation of the Cu10(p-MBT)10(PPh3)4 cluster and the emission of the Cu8(p-MBT)8(PPh3)4 cluster, combined with accurate control over the confined space between the two nanoclusters. Density functional theory is employed to provide deeper insights into the role of the distance and dipole orientations of molecules to illustrate the FRET procedure between two cluster molecules at the electronic structure level. |
| 155. |  | Roman M. Wyss, Günter Kewes, Pietro Marabotti, Stefan M. Koepfli, Karl-Philipp Schlichting, Markus Parzefall, Eric Bonvin, Martin F. Sarott, Morgan Trassin, Maximilian Oezkent, Chen-Hsun Lu, Kevin-P. Gradwohl, Thomas Perrault, Lala Habibova, Giorgia Marcelli, Marcela Giraldo, Jan Vermant, Lukas Novotny, Martin Frimmer, Mads C. Weber, Sebastian Heeg Bulk-suppressed and surface-sensitive Raman scattering by transferable plasmonic membranes with irregular slot-shaped nanopores In: Nature Communications, vol. 15, no. 5236, 2024, (Characterizing surfaces by Raman spectroscopy is limited by the competition of surface and bulk Raman responses. Here the authors use nanoporous plasmonic membranes to enhance surface Raman signals while suppressing bulk contributions.). @article{nokey,
title = {Bulk-suppressed and surface-sensitive Raman scattering by transferable plasmonic membranes with irregular slot-shaped nanopores},
author = {Roman M. Wyss and Günter Kewes and Pietro Marabotti and Stefan M. Koepfli and Karl-Philipp Schlichting and Markus Parzefall and Eric Bonvin and Martin F. Sarott and Morgan Trassin and Maximilian Oezkent and Chen-Hsun Lu and Kevin-P. Gradwohl and Thomas Perrault and Lala Habibova and Giorgia Marcelli and Marcela Giraldo and Jan Vermant and Lukas Novotny and Martin Frimmer and Mads C. Weber and Sebastian Heeg },
url = {https://www.nature.com/articles/s41467-024-49130-2.pdf},
doi = {10.1038/s41467-024-49130-2},
year = {2024},
date = {2024-06-19},
urldate = {2024-06-19},
journal = {Nature Communications},
volume = {15},
number = {5236},
abstract = {Raman spectroscopy enables the non-destructive characterization of chemical composition, crystallinity, defects, or strain in countless materials. However, the Raman response of surfaces or thin films is often weak and obscured by dominant bulk signals. Here we overcome this limitation by placing a transferable porous gold membrane, (PAuM) on the surface of interest. Slot-shaped nanopores in the membrane act as plasmonic antennas and enhance the Raman response of the surface or thin film underneath. Simultaneously, the PAuM suppresses the penetration of the excitation laser into the bulk, efficiently blocking its Raman signal. Using graphene as a model surface, we show that this method increases the surface-to-bulk Raman signal ratio by three orders of magnitude. We find that 90% of the Raman enhancement occurs within the top 2.5 nm of the material, demonstrating truly surface-sensitive Raman scattering. To validate our approach, we quantify the strain in a 12.5 nm thin Silicon film and analyze the surface of a LaNiO3 thin film. We observe a Raman mode splitting for the LaNiO3 surface-layer, which is spectroscopic evidence that the surface structure differs from the bulk. These results validate that PAuM gives direct access to Raman signatures of thin films and surfaces.},
note = {Characterizing surfaces by Raman spectroscopy is limited by the competition of surface and bulk Raman responses. Here the authors use nanoporous plasmonic membranes to enhance surface Raman signals while suppressing bulk contributions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Raman spectroscopy enables the non-destructive characterization of chemical composition, crystallinity, defects, or strain in countless materials. However, the Raman response of surfaces or thin films is often weak and obscured by dominant bulk signals. Here we overcome this limitation by placing a transferable porous gold membrane, (PAuM) on the surface of interest. Slot-shaped nanopores in the membrane act as plasmonic antennas and enhance the Raman response of the surface or thin film underneath. Simultaneously, the PAuM suppresses the penetration of the excitation laser into the bulk, efficiently blocking its Raman signal. Using graphene as a model surface, we show that this method increases the surface-to-bulk Raman signal ratio by three orders of magnitude. We find that 90% of the Raman enhancement occurs within the top 2.5 nm of the material, demonstrating truly surface-sensitive Raman scattering. To validate our approach, we quantify the strain in a 12.5 nm thin Silicon film and analyze the surface of a LaNiO3 thin film. We observe a Raman mode splitting for the LaNiO3 surface-layer, which is spectroscopic evidence that the surface structure differs from the bulk. These results validate that PAuM gives direct access to Raman signatures of thin films and surfaces. |
| 154. | 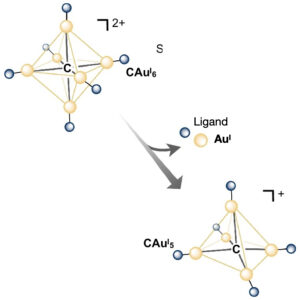 | Xiao-Li Pei, Pei Zhao, Hitoshi Ube, Zhen Lei, Masahiro Ehara, Mitsuhiko Shionoya Single-gold etching at the hypercarbon atom of C-centred hexagold(I) clusters protected by chiral N-heterocyclic carbenes In: Nature Communications, vol. 15, no. 5024, 2024, (The control of atomically precise etching of nano-sized metal clusters is important for understanding their structure-specific properties. Here, the authors report the etching of a single gold atom on a hypercarbon centre of gold(I) clusters.
). @article{nokey,
title = {Single-gold etching at the hypercarbon atom of C-centred hexagold(I) clusters protected by chiral N-heterocyclic carbenes},
author = {Xiao-Li Pei and Pei Zhao and Hitoshi Ube and Zhen Lei and Masahiro Ehara and Mitsuhiko Shionoya},
url = {https://www.nature.com/articles/s41467-024-49295-w.pdf},
doi = {10.1038/s41467-024-49295-w},
year = {2024},
date = {2024-06-12},
urldate = {2024-06-12},
journal = {Nature Communications},
volume = {15},
number = {5024},
abstract = {Chemical etching of nano-sized metal clusters at the atomic level has a high potential for creating metal number-specific structures and functions that are difficult to achieve with bottom-up synthesis methods. In particular, precisely etching metal atoms one by one from nonmetallic element-centred metal clusters and elucidating the relationship between their well-defined structures, and chemical and physical properties will facilitate future materials design for metal clusters. Here we report the single-gold etching at a hypercarbon centre in gold(I) clusters. Specifically, C-centred hexagold(I) clusters protected by chiral N-heterocyclic carbenes are etched with bisphosphine to yield C-centred pentagold(I) (CAuI5) clusters. The CAuI5 clusters exhibit an unusually large bathochromic shift in luminescence, which is reproduced theoretically. The etching mechanism is experimentally and theoretically suggested to be a tandem dissociation-association-elimination pathway. Furthermore, the vacant site of the central carbon of the CAuI5 cluster can accommodate AuCl, allowing for post-functionalisation of the C-centred gold(I) clusters.},
note = {The control of atomically precise etching of nano-sized metal clusters is important for understanding their structure-specific properties. Here, the authors report the etching of a single gold atom on a hypercarbon centre of gold(I) clusters.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chemical etching of nano-sized metal clusters at the atomic level has a high potential for creating metal number-specific structures and functions that are difficult to achieve with bottom-up synthesis methods. In particular, precisely etching metal atoms one by one from nonmetallic element-centred metal clusters and elucidating the relationship between their well-defined structures, and chemical and physical properties will facilitate future materials design for metal clusters. Here we report the single-gold etching at a hypercarbon centre in gold(I) clusters. Specifically, C-centred hexagold(I) clusters protected by chiral N-heterocyclic carbenes are etched with bisphosphine to yield C-centred pentagold(I) (CAuI5) clusters. The CAuI5 clusters exhibit an unusually large bathochromic shift in luminescence, which is reproduced theoretically. The etching mechanism is experimentally and theoretically suggested to be a tandem dissociation-association-elimination pathway. Furthermore, the vacant site of the central carbon of the CAuI5 cluster can accommodate AuCl, allowing for post-functionalisation of the C-centred gold(I) clusters. |
| 153. |  | Zhanzhao Li, Masaki Saruyama, Toru Asaka, Toshiharu Teranishi Waning-and-waxing shape changes in ionic nanoplates upon cation exchange In: Nature Communications, vol. 15, no. 4899, 2024, (The robust anion framework of ionic nanocrystals impedes shape change by cation exchange. Here, the authors report an anisotropic, regenerative transformation of Cu1.8S nanoplates during cation exchange.). @article{nokey,
title = {Waning-and-waxing shape changes in ionic nanoplates upon cation exchange},
author = {Zhanzhao Li and Masaki Saruyama and Toru Asaka and Toshiharu Teranishi },
url = {https://www.nature.com/articles/s41467-024-49294-x.pdf},
doi = {10.1038/s41467-024-49294-x},
year = {2024},
date = {2024-06-08},
urldate = {2024-06-08},
journal = {Nature Communications},
volume = {15},
number = {4899},
abstract = {Flexible control of the composition and morphology of nanocrystals (NCs) over a wide range is an essential technology for the creation of functional nanomaterials. Cation exchange (CE) is a facile method by which to finely tune the compositions of ionic NCs, providing an opportunity to obtain complex nanostructures that are difficult to form using conventional chemical synthesis procedures. However, due to their robust anion frameworks, CE cannot typically be used to modify the original morphology of the host NCs. In this study, we report an anisotropic morphological transformation of Cu1.8S NCs during CE. Upon partial CE of Cu1.8S nanoplates (NPLs) with Mn2+, the hexagonal NPLs are transformed into crescent-shaped Cu1.8S–MnS NPLs. Upon further CE, these crescent-shaped NPLs evolve back into completely hexagonal MnS NPLs. Comprehensive characterization of the intermediates reveals that this waxing-and-waning shape-evolution process is due to dissolution, redeposition, and intraparticle migration of Cu+ and S2−. Furthermore, in addition to Mn2+, this CE-induced transformation process occurs with Zn2+, Cd2+ and Fe3+. This finding presents a strategy by which to create heterostructured NCs with various morphologies and compositions under mild conditions.},
note = {The robust anion framework of ionic nanocrystals impedes shape change by cation exchange. Here, the authors report an anisotropic, regenerative transformation of Cu1.8S nanoplates during cation exchange.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Flexible control of the composition and morphology of nanocrystals (NCs) over a wide range is an essential technology for the creation of functional nanomaterials. Cation exchange (CE) is a facile method by which to finely tune the compositions of ionic NCs, providing an opportunity to obtain complex nanostructures that are difficult to form using conventional chemical synthesis procedures. However, due to their robust anion frameworks, CE cannot typically be used to modify the original morphology of the host NCs. In this study, we report an anisotropic morphological transformation of Cu1.8S NCs during CE. Upon partial CE of Cu1.8S nanoplates (NPLs) with Mn2+, the hexagonal NPLs are transformed into crescent-shaped Cu1.8S–MnS NPLs. Upon further CE, these crescent-shaped NPLs evolve back into completely hexagonal MnS NPLs. Comprehensive characterization of the intermediates reveals that this waxing-and-waning shape-evolution process is due to dissolution, redeposition, and intraparticle migration of Cu+ and S2−. Furthermore, in addition to Mn2+, this CE-induced transformation process occurs with Zn2+, Cd2+ and Fe3+. This finding presents a strategy by which to create heterostructured NCs with various morphologies and compositions under mild conditions. |
| 152. |  | Huacheng Li, Xin Xu, Rongcheng Guan, Artur Movsesyan, Zhenni Lu, Qiliang Xu, Ziyun Jiang, Yurong Yang, Majid Khan, Jin Wen, Hongwei Wu, Santiago de la Moya, Gil Markovich, Huatian Hu, Zhiming Wang, Qiang Guo, Tao Yi, Alexander O. Govorov, Zhiyong Tang, Xiang Lan Collective chiroptical activity through the interplay of excitonic and charge-transfer effects in localized plasmonic fields In: Nature Communications, vol. 15, no. 4846, 2024, (Chiral interaction of molecular ensembles with confined light fields is elusive. Here, the authors report collective chiroptical effects through coupling of exciton and charge-transfer mixed molecular assemblies with plasmonic nanoparticles.). @article{nokey,
title = {Collective chiroptical activity through the interplay of excitonic and charge-transfer effects in localized plasmonic fields},
author = {Huacheng Li and Xin Xu and Rongcheng Guan and Artur Movsesyan and Zhenni Lu and Qiliang Xu and Ziyun Jiang and Yurong Yang and Majid Khan and Jin Wen and Hongwei Wu and Santiago de la Moya and Gil Markovich and Huatian Hu and Zhiming Wang and Qiang Guo and Tao Yi and Alexander O. Govorov and Zhiyong Tang and Xiang Lan },
url = {https://www.nature.com/articles/s41467-024-49086-3.pdf},
doi = {10.1038/s41467-024-49086-3},
year = {2024},
date = {2024-06-06},
urldate = {2024-06-06},
journal = {Nature Communications},
volume = {15},
number = {4846},
abstract = {The collective light-matter interaction of chiral supramolecular aggregates or molecular ensembles with confined light fields remains a mystery beyond the current theoretical description. Here, we programmably and accurately build models of chiral plasmonic complexes, aiming to uncover the entangled effects of excitonic correlations, intra- and intermolecular charge transfer, and localized surface plasmon resonances. The intricate interplay of multiple chirality origins has proven to be strongly dependent on the site-specificity of chiral molecules on plasmonic nanoparticle surfaces spanning the nanometer to sub-nanometer scale. This dependence is manifested as a distinct circular dichroism response that varies in spectral asymmetry/splitting, signal intensity, and internal ratio of intensity. The inhomogeneity of the surface-localized plasmonic field is revealed to affect excitonic and charge-transfer mixed intermolecular couplings, which are inherent to chirality generation and amplification. Our findings contribute to the development of hybrid classical-quantum theoretical frameworks and the harnessing of spin-charge transport for emergent applications.},
note = {Chiral interaction of molecular ensembles with confined light fields is elusive. Here, the authors report collective chiroptical effects through coupling of exciton and charge-transfer mixed molecular assemblies with plasmonic nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The collective light-matter interaction of chiral supramolecular aggregates or molecular ensembles with confined light fields remains a mystery beyond the current theoretical description. Here, we programmably and accurately build models of chiral plasmonic complexes, aiming to uncover the entangled effects of excitonic correlations, intra- and intermolecular charge transfer, and localized surface plasmon resonances. The intricate interplay of multiple chirality origins has proven to be strongly dependent on the site-specificity of chiral molecules on plasmonic nanoparticle surfaces spanning the nanometer to sub-nanometer scale. This dependence is manifested as a distinct circular dichroism response that varies in spectral asymmetry/splitting, signal intensity, and internal ratio of intensity. The inhomogeneity of the surface-localized plasmonic field is revealed to affect excitonic and charge-transfer mixed intermolecular couplings, which are inherent to chirality generation and amplification. Our findings contribute to the development of hybrid classical-quantum theoretical frameworks and the harnessing of spin-charge transport for emergent applications. |
| 151. |  | Olga Guselnikova, Andrii Trelin, Yunqing Kang, Pavel Postnikov, Makoto Kobashi, Asuka Suzuki, Lok Kumar Shrestha, Joel Henzie, Yusuke Yamauchi Pretreatment-free SERS sensing of microplastics using a self-attention-based neural network on hierarchically porous Ag foams In: Nature Communications, vol. 15, no. 4351, 2024, (Detection and identification of microplastics (MPs) in environmental samples is hampered by the need for isolation and pretreatment methods. Here, the authors combine porous Ag substrates with self-attention neural networks to directly identify six types of MPs in environmental samples.
). @article{nokey,
title = {Pretreatment-free SERS sensing of microplastics using a self-attention-based neural network on hierarchically porous Ag foams},
author = {Olga Guselnikova and Andrii Trelin and Yunqing Kang and Pavel Postnikov and Makoto Kobashi and Asuka Suzuki and Lok Kumar Shrestha and Joel Henzie and Yusuke Yamauchi },
url = {https://www.nature.com/articles/s41467-024-48148-w.pdf},
doi = {10.1038/s41467-024-48148-w},
year = {2024},
date = {2024-05-28},
urldate = {2024-05-28},
journal = {Nature Communications},
volume = {15},
number = {4351},
abstract = {Low-cost detection systems are needed for the identification of microplastics (MPs) in environmental samples. However, their rapid identification is hindered by the need for complex isolation and pre-treatment methods. This study describes a comprehensive sensing platform to identify MPs in environmental samples without requiring independent separation or pre-treatment protocols. It leverages the physicochemical properties of macroporous-mesoporous silver (Ag) substrates templated with self-assembled polymeric micelles to concurrently separate and analyze multiple MP targets using surface-enhanced Raman spectroscopy (SERS). The hydrophobic layer on Ag aids in stabilizing the nanostructures in the environment and mitigates biofouling. To monitor complex samples with multiple MPs and to demultiplex numerous overlapping patterns, we develop a neural network (NN) algorithm called SpecATNet that employs a self-attention mechanism to resolve the complex dependencies and patterns in SERS data to identify six common types of MPs: polystyrene, polyethylene, polymethylmethacrylate, polytetrafluoroethylene, nylon, and polyethylene terephthalate. SpecATNet uses multi-label classification to analyze multi-component mixtures even in the presence of various interference agents. The combination of macroporous-mesoporous Ag substrates and self-attention-based NN technology holds potential to enable field monitoring of MPs by generating rich datasets that machines can interpret and analyze.},
note = {Detection and identification of microplastics (MPs) in environmental samples is hampered by the need for isolation and pretreatment methods. Here, the authors combine porous Ag substrates with self-attention neural networks to directly identify six types of MPs in environmental samples.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Low-cost detection systems are needed for the identification of microplastics (MPs) in environmental samples. However, their rapid identification is hindered by the need for complex isolation and pre-treatment methods. This study describes a comprehensive sensing platform to identify MPs in environmental samples without requiring independent separation or pre-treatment protocols. It leverages the physicochemical properties of macroporous-mesoporous silver (Ag) substrates templated with self-assembled polymeric micelles to concurrently separate and analyze multiple MP targets using surface-enhanced Raman spectroscopy (SERS). The hydrophobic layer on Ag aids in stabilizing the nanostructures in the environment and mitigates biofouling. To monitor complex samples with multiple MPs and to demultiplex numerous overlapping patterns, we develop a neural network (NN) algorithm called SpecATNet that employs a self-attention mechanism to resolve the complex dependencies and patterns in SERS data to identify six common types of MPs: polystyrene, polyethylene, polymethylmethacrylate, polytetrafluoroethylene, nylon, and polyethylene terephthalate. SpecATNet uses multi-label classification to analyze multi-component mixtures even in the presence of various interference agents. The combination of macroporous-mesoporous Ag substrates and self-attention-based NN technology holds potential to enable field monitoring of MPs by generating rich datasets that machines can interpret and analyze. |
| 150. |  | Xiaokang Yao, Yuxin Li, Huifang Shi, Ze Yu, Beishen Wu, Zixing Zhou, Chifeng Zhou, Xifang Zheng, Mengting Tang, Xiao Wang, Huili Ma, Zhengong Meng, Wei Huang, Zhongfu An Narrowband room temperature phosphorescence of closed-loop molecules through the multiple resonance effect In: Nature Communications, vol. 15, no. 4520, 2024, (Luminescent materials with narrowband emissions are vital for optoelectronic applications. Here, the authors achieve room temperature phosphorescence with a FWHM of 30 nm through the multiple resonance effect and showcase its practical application in X-ray imaging.). @article{nokey,
title = {Narrowband room temperature phosphorescence of closed-loop molecules through the multiple resonance effect},
author = {Xiaokang Yao and Yuxin Li and Huifang Shi and Ze Yu and Beishen Wu and Zixing Zhou and Chifeng Zhou and Xifang Zheng and Mengting Tang and Xiao Wang and Huili Ma and Zhengong Meng and Wei Huang and Zhongfu An },
url = {https://www.nature.com/articles/s41467-024-48856-3.pdf},
doi = {10.1038/s41467-024-48856-3},
year = {2024},
date = {2024-05-28},
urldate = {2024-05-28},
journal = {Nature Communications},
volume = {15},
number = {4520},
abstract = {Luminescent materials with narrowband emission show great potential for diverse applications in optoelectronics. Purely organic phosphors with room-temperature phosphorescence (RTP) have made significant success in rationally manipulating quantum efficiency, lifetimes, and colour gamut in the past years, but there is limited attention on the purity of the RTP colours. Herein we report a series of closed-loop molecules with narrowband phosphorescence by multiple resonance effect, which significantly improves the colour purity of RTP. Phosphors show narrowband phosphorescence with full width at half maxima (FWHM) of 30 nm after doping into a rigid benzophenone matrix under ambient conditions, of which the RTP efficiency reaches 51.8%. At 77 K, the FWHM of phosphorescence is only 11 nm. Meanwhile, the colour of narrowband RTP can be tuned from sky blue to green with the modification of methyl groups. Additionally, the potential applications in X-ray imaging and display are demonstrated. This work not only outlines a design principle for developing narrowband RTP materials but also makes a major step forward extending the potential applications of narrowband luminescent materials in optoelectronics.},
note = {Luminescent materials with narrowband emissions are vital for optoelectronic applications. Here, the authors achieve room temperature phosphorescence with a FWHM of 30 nm through the multiple resonance effect and showcase its practical application in X-ray imaging.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Luminescent materials with narrowband emission show great potential for diverse applications in optoelectronics. Purely organic phosphors with room-temperature phosphorescence (RTP) have made significant success in rationally manipulating quantum efficiency, lifetimes, and colour gamut in the past years, but there is limited attention on the purity of the RTP colours. Herein we report a series of closed-loop molecules with narrowband phosphorescence by multiple resonance effect, which significantly improves the colour purity of RTP. Phosphors show narrowband phosphorescence with full width at half maxima (FWHM) of 30 nm after doping into a rigid benzophenone matrix under ambient conditions, of which the RTP efficiency reaches 51.8%. At 77 K, the FWHM of phosphorescence is only 11 nm. Meanwhile, the colour of narrowband RTP can be tuned from sky blue to green with the modification of methyl groups. Additionally, the potential applications in X-ray imaging and display are demonstrated. This work not only outlines a design principle for developing narrowband RTP materials but also makes a major step forward extending the potential applications of narrowband luminescent materials in optoelectronics. |
| 149. |  | Yufei Wang, Yilong Zhou, Quanpeng Yang, Rourav Basak, Yu Xie, Dong Le, Alexander D. Fuqua, Wade Shipley, Zachary Yam, Alex Frano, Gaurav Arya, Andrea R. Tao Self-assembly of nanocrystal checkerboard patterns via non-specific interactions In: Nature Communications, vol. 15, no. 3913, 2024, (The self-assembly of nanocrystals into checkerboard lattice patterns is difficult to control. Here, the authors investigate the formation of such patterns from hydrophilic/hydrophobic bifunctionalized Ag nanocubes and use multiscale simulations to understand the effects of physical forces.). @article{nokey,
title = {Self-assembly of nanocrystal checkerboard patterns via non-specific interactions},
author = {Yufei Wang and Yilong Zhou and Quanpeng Yang and Rourav Basak and Yu Xie and Dong Le and Alexander D. Fuqua and Wade Shipley and Zachary Yam and Alex Frano and Gaurav Arya and Andrea R. Tao },
url = {https://www.nature.com/articles/s41467-024-47572-2.pdf},
doi = {10.1038/s41467-024-47572-2},
year = {2024},
date = {2024-05-09},
urldate = {2024-05-09},
journal = {Nature Communications},
volume = {15},
number = {3913},
abstract = {Checkerboard lattices—where the resulting structure is open, porous, and highly symmetric—are difficult to create by self-assembly. Synthetic systems that adopt such structures typically rely on shape complementarity and site-specific chemical interactions that are only available to biomolecular systems (e.g., protein, DNA). Here we show the assembly of checkerboard lattices from colloidal nanocrystals that harness the effects of multiple, coupled physical forces at disparate length scales (interfacial, interparticle, and intermolecular) and that do not rely on chemical binding. Colloidal Ag nanocubes were bi-functionalized with mixtures of hydrophilic and hydrophobic surface ligands and subsequently assembled at an air–water interface. Using feedback between molecular dynamics simulations and interfacial assembly experiments, we achieve a periodic checkerboard mesostructure that represents a tiny fraction of the phase space associated with the polymer-grafted nanocrystals used in these experiments. In a broader context, this work expands our knowledge of non-specific nanocrystal interactions and presents a computation-guided strategy for designing self-assembling materials.},
note = {The self-assembly of nanocrystals into checkerboard lattice patterns is difficult to control. Here, the authors investigate the formation of such patterns from hydrophilic/hydrophobic bifunctionalized Ag nanocubes and use multiscale simulations to understand the effects of physical forces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Checkerboard lattices—where the resulting structure is open, porous, and highly symmetric—are difficult to create by self-assembly. Synthetic systems that adopt such structures typically rely on shape complementarity and site-specific chemical interactions that are only available to biomolecular systems (e.g., protein, DNA). Here we show the assembly of checkerboard lattices from colloidal nanocrystals that harness the effects of multiple, coupled physical forces at disparate length scales (interfacial, interparticle, and intermolecular) and that do not rely on chemical binding. Colloidal Ag nanocubes were bi-functionalized with mixtures of hydrophilic and hydrophobic surface ligands and subsequently assembled at an air–water interface. Using feedback between molecular dynamics simulations and interfacial assembly experiments, we achieve a periodic checkerboard mesostructure that represents a tiny fraction of the phase space associated with the polymer-grafted nanocrystals used in these experiments. In a broader context, this work expands our knowledge of non-specific nanocrystal interactions and presents a computation-guided strategy for designing self-assembling materials. |
| 148. |  | Hong Kang, Yuexuan Yang, Bryan Wei Synthetic molecular switches driven by DNA-modifying enzymes In: Nature Communications, vol. 15, no. 3781, 2024, (Molecular switches are ubiquitous in the biochemistry regulatory network. Here, the authors construct synthetic molecular switches controlled by DNA-modifying enzymes such as DNA polymerase and nicking endonuclease to control and cascade assembly and disassembly.). @article{nokey,
title = {Synthetic molecular switches driven by DNA-modifying enzymes},
author = {Hong Kang and Yuexuan Yang and Bryan Wei },
url = {https://www.nature.com/articles/s41467-024-47742-2.pdf},
doi = {10.1038/s41467-024-47742-2},
year = {2024},
date = {2024-05-06},
urldate = {2024-05-06},
journal = {Nature Communications},
volume = {15},
number = {3781},
abstract = {Taking inspiration from natural systems, in which molecular switches are ubiquitous in the biochemistry regulatory network, we aim to design and construct synthetic molecular switches driven by DNA-modifying enzymes, such as DNA polymerase and nicking endonuclease. The enzymatic treatments on our synthetic DNA constructs controllably switch ON or OFF the sticky end cohesion and in turn cascade to the structural association or disassociation. Here we showcase the concept in multiple DNA nanostructure systems with robust assembly/disassembly performance. The switch mechanisms are first illustrated in minimalist systems with a few DNA strands. Then the ON/OFF switches are realized in complex DNA lattice and origami systems with designated morphological changes responsive to the specific enzymatic treatments.},
note = {Molecular switches are ubiquitous in the biochemistry regulatory network. Here, the authors construct synthetic molecular switches controlled by DNA-modifying enzymes such as DNA polymerase and nicking endonuclease to control and cascade assembly and disassembly.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Taking inspiration from natural systems, in which molecular switches are ubiquitous in the biochemistry regulatory network, we aim to design and construct synthetic molecular switches driven by DNA-modifying enzymes, such as DNA polymerase and nicking endonuclease. The enzymatic treatments on our synthetic DNA constructs controllably switch ON or OFF the sticky end cohesion and in turn cascade to the structural association or disassociation. Here we showcase the concept in multiple DNA nanostructure systems with robust assembly/disassembly performance. The switch mechanisms are first illustrated in minimalist systems with a few DNA strands. Then the ON/OFF switches are realized in complex DNA lattice and origami systems with designated morphological changes responsive to the specific enzymatic treatments. |
| 147. |  | Cameron J. Owen, Yu Xie, Anders Johansson, Lixin Sun, Boris Kozinsky Low-index mesoscopic surface reconstructions of Au surfaces using Bayesian force fields In: Nature Communications, vol. 15, no. 3790, 2024, (Metal surfaces have long been known to reconstruct, but key mechanistic aspects are poorly understood. Here, the authors use Bayesian force fields to gain insights into gold surface reconstructions that are crucial for material science and catalysis.). @article{nokey,
title = {Low-index mesoscopic surface reconstructions of Au surfaces using Bayesian force fields},
author = {Cameron J. Owen and Yu Xie and Anders Johansson and Lixin Sun and Boris Kozinsky },
url = {https://www.nature.com/articles/s41467-024-48192-6.pdf},
doi = {10.1038/s41467-024-48192-6},
year = {2024},
date = {2024-05-06},
urldate = {2024-05-06},
journal = {Nature Communications},
volume = {15},
number = {3790},
abstract = {Metal surfaces have long been known to reconstruct, significantly influencing their structural and catalytic properties. Many key mechanistic aspects of these subtle transformations remain poorly understood due to limitations of previous simulation approaches. Using active learning of Bayesian machine-learned force fields trained from ab initio calculations, we enable large-scale molecular dynamics simulations to describe the thermodynamics and time evolution of the low-index mesoscopic surface reconstructions of Au (e.g., the Au(111)-‘Herringbone,’ Au(110)-(1 × 2)-‘Missing-Row,’ and Au(100)-‘Quasi-Hexagonal’ reconstructions). This capability yields direct atomistic understanding of the dynamic emergence of these surface states from their initial facets, providing previously inaccessible information such as nucleation kinetics and a complete mechanistic interpretation of reconstruction under the effects of strain and local deviations from the original stoichiometry. We successfully reproduce previous experimental observations of reconstructions on pristine surfaces and provide quantitative predictions of the emergence of spinodal decomposition and localized reconstruction in response to strain at non-ideal stoichiometries. A unified mechanistic explanation is presented of the kinetic and thermodynamic factors driving surface reconstruction. Furthermore, we study surface reconstructions on Au nanoparticles, where characteristic (111) and (100) reconstructions spontaneously appear on a variety of high-symmetry particle morphologies.},
note = {Metal surfaces have long been known to reconstruct, but key mechanistic aspects are poorly understood. Here, the authors use Bayesian force fields to gain insights into gold surface reconstructions that are crucial for material science and catalysis.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Metal surfaces have long been known to reconstruct, significantly influencing their structural and catalytic properties. Many key mechanistic aspects of these subtle transformations remain poorly understood due to limitations of previous simulation approaches. Using active learning of Bayesian machine-learned force fields trained from ab initio calculations, we enable large-scale molecular dynamics simulations to describe the thermodynamics and time evolution of the low-index mesoscopic surface reconstructions of Au (e.g., the Au(111)-‘Herringbone,’ Au(110)-(1 × 2)-‘Missing-Row,’ and Au(100)-‘Quasi-Hexagonal’ reconstructions). This capability yields direct atomistic understanding of the dynamic emergence of these surface states from their initial facets, providing previously inaccessible information such as nucleation kinetics and a complete mechanistic interpretation of reconstruction under the effects of strain and local deviations from the original stoichiometry. We successfully reproduce previous experimental observations of reconstructions on pristine surfaces and provide quantitative predictions of the emergence of spinodal decomposition and localized reconstruction in response to strain at non-ideal stoichiometries. A unified mechanistic explanation is presented of the kinetic and thermodynamic factors driving surface reconstruction. Furthermore, we study surface reconstructions on Au nanoparticles, where characteristic (111) and (100) reconstructions spontaneously appear on a variety of high-symmetry particle morphologies. |
| 146. |  | Xue-Guang Chen, Linhan Lin, Guan-Yao Huang, Xiao-Mei Chen, Xiao-Ze Li, Yun-Ke Zhou, Yixuan Zou, Tairan Fu, Peng Li, Zhengcao Li, Hong-Bo Sun Optofluidic crystallithography for directed growth of single-crystalline halide perovskites In: Nature Communications, vol. 15, no. 3677, 2024, (Precise and spatio-temporal control of crystallization kinetics is important but challenging. Here, the authors propose an optical strategy called optofluidic crystallithography to steer the growth of single-crystalline halide perovskites.). @article{nokey,
title = {Optofluidic crystallithography for directed growth of single-crystalline halide perovskites},
author = {Xue-Guang Chen and Linhan Lin and Guan-Yao Huang and Xiao-Mei Chen and Xiao-Ze Li and Yun-Ke Zhou and Yixuan Zou and Tairan Fu and Peng Li and Zhengcao Li and Hong-Bo Sun },
url = {https://www.nature.com/articles/s41467-024-48110-w.pdf},
doi = {10.1038/s41467-024-48110-w},
year = {2024},
date = {2024-05-01},
urldate = {2024-05-01},
journal = {Nature Communications},
volume = {15},
number = {3677},
abstract = {Crystallization is a fundamental phenomenon which describes how the atomic building blocks such as atoms and molecules are arranged into ordered or quasi-ordered structure and form solid-state materials. While numerous studies have focused on the nucleation behavior, the precise and spatiotemporal control of growth kinetics, which dictates the defect density, the micromorphology, as well as the properties of the grown materials, remains elusive so far. Herein, we propose an optical strategy, termed optofluidic crystallithography (OCL), to solve this fundamental problem. Taking halide perovskites as an example, we use a laser beam to manipulate the molecular motion in the native precursor environment and create inhomogeneous spatial distribution of the molecular species. Harnessing the coordinated effect of laser-controlled local supersaturation and interfacial energy, we precisely steer the ionic reaction at the growth interface and directly print arbitrary single crystals of halide perovskites of high surface quality, crystallinity, and uniformity at a high printing speed of 102 μm s−1. The OCL technique can be potentially extended to the fabrication of single-crystal structures beyond halide perovskites, once crystallization can be triggered under the laser-directed local supersaturation.},
note = {Precise and spatio-temporal control of crystallization kinetics is important but challenging. Here, the authors propose an optical strategy called optofluidic crystallithography to steer the growth of single-crystalline halide perovskites.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Crystallization is a fundamental phenomenon which describes how the atomic building blocks such as atoms and molecules are arranged into ordered or quasi-ordered structure and form solid-state materials. While numerous studies have focused on the nucleation behavior, the precise and spatiotemporal control of growth kinetics, which dictates the defect density, the micromorphology, as well as the properties of the grown materials, remains elusive so far. Herein, we propose an optical strategy, termed optofluidic crystallithography (OCL), to solve this fundamental problem. Taking halide perovskites as an example, we use a laser beam to manipulate the molecular motion in the native precursor environment and create inhomogeneous spatial distribution of the molecular species. Harnessing the coordinated effect of laser-controlled local supersaturation and interfacial energy, we precisely steer the ionic reaction at the growth interface and directly print arbitrary single crystals of halide perovskites of high surface quality, crystallinity, and uniformity at a high printing speed of 102 μm s−1. The OCL technique can be potentially extended to the fabrication of single-crystal structures beyond halide perovskites, once crystallization can be triggered under the laser-directed local supersaturation. |
| 145. |  | Danman Guo, Wen Wang, Kaimin Zhang, Jinzheng Chen, Yuyuan Wang, Tianyi Wang, Wangmeng Hou, Zhen Zhang, Huahua Huang, Zhenguo Chi, Zhiyong Yang Visible-light-excited robust room-temperature phosphorescence of dimeric single-component luminophores in the amorphous state In: Nature Communications, vol. 15, no. 3598, 2024, (Organic room-temperature phosphorescence (RTP) is limited to rigid environments. Here, the authors report a single-component system with robust persistent RTP emissions in various aggregation states, such as crystalline, fine powder, and amorphous.). @article{nokey,
title = {Visible-light-excited robust room-temperature phosphorescence of dimeric single-component luminophores in the amorphous state},
author = {Danman Guo and Wen Wang and Kaimin Zhang and Jinzheng Chen and Yuyuan Wang and Tianyi Wang and Wangmeng Hou and Zhen Zhang and Huahua Huang and Zhenguo Chi and Zhiyong Yang },
url = {https://www.nature.com/articles/s41467-024-47937-7.pdf},
doi = {10.1038/s41467-024-47937-7},
year = {2024},
date = {2024-04-27},
urldate = {2024-04-27},
journal = {Nature Communications},
volume = {15},
number = {3598},
abstract = {Organic room temperature phosphorescence (RTP) has significant potential in various applications of information storage, anti-counterfeiting, and bio-imaging. However, achieving robust organic RTP emission of the single-component system is challenging to overcome the restriction of the crystalline state or other rigid environments with cautious treatment. Herein, we report a single-component system with robust persistent RTP emission in various aggregated forms, such as crystal, fine powder, and even amorphous states. Our experimental data reveal that the vigorous RTP emissions rely on their tight dimers based on strong and large-overlap π-π interactions between polycyclic aromatic hydrocarbon (PAH) groups. The dimer structure can offer not only excitons in low energy levels for visible-light excited red long-lived RTP but also suppression of the nonradiative decays even in an amorphous state for good resistance of RTP to heat (up to 70 °C) or water. Furthermore, we demonstrate the water-dispersible nanoparticle with persistent RTP over 600 nm and a lifetime of 0.22 s for visible-light excited cellular and in-vivo imaging, prepared through the common microemulsion approach without overcaution for nanocrystal formation.},
note = {Organic room-temperature phosphorescence (RTP) is limited to rigid environments. Here, the authors report a single-component system with robust persistent RTP emissions in various aggregation states, such as crystalline, fine powder, and amorphous.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Organic room temperature phosphorescence (RTP) has significant potential in various applications of information storage, anti-counterfeiting, and bio-imaging. However, achieving robust organic RTP emission of the single-component system is challenging to overcome the restriction of the crystalline state or other rigid environments with cautious treatment. Herein, we report a single-component system with robust persistent RTP emission in various aggregated forms, such as crystal, fine powder, and even amorphous states. Our experimental data reveal that the vigorous RTP emissions rely on their tight dimers based on strong and large-overlap π-π interactions between polycyclic aromatic hydrocarbon (PAH) groups. The dimer structure can offer not only excitons in low energy levels for visible-light excited red long-lived RTP but also suppression of the nonradiative decays even in an amorphous state for good resistance of RTP to heat (up to 70 °C) or water. Furthermore, we demonstrate the water-dispersible nanoparticle with persistent RTP over 600 nm and a lifetime of 0.22 s for visible-light excited cellular and in-vivo imaging, prepared through the common microemulsion approach without overcaution for nanocrystal formation. |
| 144. |  | Cheng Hu, Jiajun Chen, Xianliang Zhou, Yufeng Xie, Xinyue Huang, Zhenghan Wu, Saiqun Ma, Zhichun Zhang, Kunqi Xu, Neng Wan, Yueheng Zhang, Qi Liang, Zhiwen Shi Collapse of carbon nanotubes due to local high-pressure from van der Waals encapsulation In: Nature Communications, vol. 15, no. 3486, 2024, (Van der Waals (vdW) assembly of low-dimensional materials has proven the capability of creating structures with on-demand properties. Here, the authors report on the structural collapse of CNTs in conjunction with a metal-semiconductor junction induced by the VdW encapsulation.). @article{nokey,
title = {Collapse of carbon nanotubes due to local high-pressure from van der Waals encapsulation},
author = {Cheng Hu and Jiajun Chen and Xianliang Zhou and Yufeng Xie and Xinyue Huang and Zhenghan Wu and Saiqun Ma and Zhichun Zhang and Kunqi Xu and Neng Wan and Yueheng Zhang and Qi Liang and Zhiwen Shi },
url = {https://www.nature.com/articles/s41467-024-47903-3.pdf},
doi = {10.1038/s41467-024-47903-3},
year = {2024},
date = {2024-04-25},
urldate = {2024-04-25},
journal = {Nature Communications},
volume = {15},
number = {3486},
abstract = {Van der Waals (vdW) assembly of low-dimensional materials has proven the capability of creating structures with on-demand properties. It is predicted that the vdW encapsulation can induce a local high-pressure of a few GPa, which will strongly modify the structure and property of trapped materials. Here, we report on the structural collapse of carbon nanotubes (CNTs) induced by the vdW encapsulation. By simply covering CNTs with a hexagonal boron nitride flake, most of the CNTs (≈77%) convert from a tubular structure to a collapsed flat structure. Regardless of their original diameters, all the collapsed CNTs exhibit a uniform height of ≈0.7 nm, which is roughly the thickness of bilayer graphene. Such structural collapse is further confirmed by Raman spectroscopy, which shows a prominent broadening and blue shift in the Raman G-peak. The vdW encapsulation-induced collapse of CNTs is fully captured by molecular dynamics simulations of the local vdW pressure. Further near-field optical characterization reveals a metal-semiconductor transition in accompany with the CNT structural collapse. Our study provides not only a convenient approach to generate local high-pressure for fundamental research, but also a collapsed-CNT semiconductor for nanoelectronic applications.},
note = {Van der Waals (vdW) assembly of low-dimensional materials has proven the capability of creating structures with on-demand properties. Here, the authors report on the structural collapse of CNTs in conjunction with a metal-semiconductor junction induced by the VdW encapsulation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Van der Waals (vdW) assembly of low-dimensional materials has proven the capability of creating structures with on-demand properties. It is predicted that the vdW encapsulation can induce a local high-pressure of a few GPa, which will strongly modify the structure and property of trapped materials. Here, we report on the structural collapse of carbon nanotubes (CNTs) induced by the vdW encapsulation. By simply covering CNTs with a hexagonal boron nitride flake, most of the CNTs (≈77%) convert from a tubular structure to a collapsed flat structure. Regardless of their original diameters, all the collapsed CNTs exhibit a uniform height of ≈0.7 nm, which is roughly the thickness of bilayer graphene. Such structural collapse is further confirmed by Raman spectroscopy, which shows a prominent broadening and blue shift in the Raman G-peak. The vdW encapsulation-induced collapse of CNTs is fully captured by molecular dynamics simulations of the local vdW pressure. Further near-field optical characterization reveals a metal-semiconductor transition in accompany with the CNT structural collapse. Our study provides not only a convenient approach to generate local high-pressure for fundamental research, but also a collapsed-CNT semiconductor for nanoelectronic applications. |
| 143. | 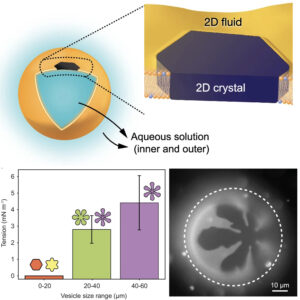 | Hao Wan, Geunwoong Jeon, Weiyue Xin, Gregory M. Grason, Maria M. Santore Flower-shaped 2D crystals grown in curved fluid vesicle membranes In: Nature Communications, vol. 15, no. 3442, 2024, (Thin crystals grown on rigid spherical templates of increasing curvature exhibit increased protrusions. Here, the authors demonstrate the opposite curvature effect on the morphology of molecularly thin crystals grown within elastic fluid membranes, like those of biological cells.). @article{nokey,
title = {Flower-shaped 2D crystals grown in curved fluid vesicle membranes},
author = {Hao Wan and Geunwoong Jeon and Weiyue Xin and Gregory M. Grason and Maria M. Santore },
url = {https://www.nature.com/articles/s41467-024-47844-x.pdf},
doi = {10.1038/s41467-024-47844-x},
year = {2024},
date = {2024-04-24},
urldate = {2024-04-24},
journal = {Nature Communications},
volume = {15},
number = {3442},
abstract = {The morphologies of two-dimensional (2D) crystals, nucleated, grown, and integrated within 2D elastic fluids, for instance in giant vesicle membranes, are dictated by an interplay of mechanics, permeability, and thermal contraction. Mitigation of solid strain drives the formation of crystals with vanishing Gaussian curvature (i.e., developable domain shapes) and, correspondingly, enhanced Gaussian curvature in the surrounding 2D fluid. However, upon cooling to grow the crystals, large vesicles sustain greater inflation and tension because their small area-to-volume ratio slows water permeation. As a result, more elaborate shapes, for instance, flowers with bendable but inextensible petals, form on large vesicles despite their more gradual curvature, while small vesicles harbor compact planar crystals. This size dependence runs counter to the known cumulative growth of strain energy of 2D colloidal crystals on rigid spherical templates. This interplay of intra-membrane mechanics and processing points to the scalable production of flexible molecular crystals of controllable complex shape.},
note = {Thin crystals grown on rigid spherical templates of increasing curvature exhibit increased protrusions. Here, the authors demonstrate the opposite curvature effect on the morphology of molecularly thin crystals grown within elastic fluid membranes, like those of biological cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The morphologies of two-dimensional (2D) crystals, nucleated, grown, and integrated within 2D elastic fluids, for instance in giant vesicle membranes, are dictated by an interplay of mechanics, permeability, and thermal contraction. Mitigation of solid strain drives the formation of crystals with vanishing Gaussian curvature (i.e., developable domain shapes) and, correspondingly, enhanced Gaussian curvature in the surrounding 2D fluid. However, upon cooling to grow the crystals, large vesicles sustain greater inflation and tension because their small area-to-volume ratio slows water permeation. As a result, more elaborate shapes, for instance, flowers with bendable but inextensible petals, form on large vesicles despite their more gradual curvature, while small vesicles harbor compact planar crystals. This size dependence runs counter to the known cumulative growth of strain energy of 2D colloidal crystals on rigid spherical templates. This interplay of intra-membrane mechanics and processing points to the scalable production of flexible molecular crystals of controllable complex shape. |
| 142. |  | Liangliang Cai, Tianhao Gao, Andrew T. S. Wee Topology selectivity of a conformationally flexible precursor through selenium doping In: Nature Communications, vol. 15, no. 3235, 2024, (Research into the control of conformational arrangements is of great importance for achieving bespoke nanoarchitectures. Here, the authors achieve topology selectivity of a conformationally flexible precursor by Se doping.). @article{nokey,
title = {Topology selectivity of a conformationally flexible precursor through selenium doping},
author = {Liangliang Cai and Tianhao Gao and Andrew T. S. Wee },
url = {https://www.nature.com/articles/s41467-024-47614-9.pdf},
doi = {10.1038/s41467-024-47614-9},
year = {2024},
date = {2024-04-15},
urldate = {2024-04-15},
journal = {Nature Communications},
volume = {15},
number = {3235},
abstract = {Conformational arrangements within nanostructures play a crucial role in shaping the overall configuration and determining the properties, for example in covalent/metal organic frameworks. In on-surface synthesis, conformational diversity often leads to uncontrollable or disordered structures. Therefore, the exploration of controlling and directing the conformational arrangements is significant in achieving desired nanoarchitectures. Herein, a conformationally flexible precursor 2,4,6-tris(3-bromophenyl)−1,3,5-triazine is employed, and a random phase consisting of C3h and Cs conformers is firstly obtained after deposition of the precursor on Cu(111) at room temperature to 365 K. At low coverage (0.01 ML) selenium doping, we achieve the selectivity of the C3h conformer and improve the nanopore structural homogeneity. The ordered two-dimensional metal organic nanostructure can be fulfilled by selenium doping from room temperature to 365 K. The formation of the conformationally flexible precursor on Cu(111) is explored through the combination of high-resolution scanning tunneling microscopy and non-contact atomic force microscopy. The regulation of energy diagrams in the absence or presence of the Se atom is revealed by density functional theory calculations. These results can enrich the on-surface synthesis toolbox of conformationally flexible precursors, for the design of complex nanoarchitectures, and for future development of engineered nanomaterials.},
note = {Research into the control of conformational arrangements is of great importance for achieving bespoke nanoarchitectures. Here, the authors achieve topology selectivity of a conformationally flexible precursor by Se doping.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Conformational arrangements within nanostructures play a crucial role in shaping the overall configuration and determining the properties, for example in covalent/metal organic frameworks. In on-surface synthesis, conformational diversity often leads to uncontrollable or disordered structures. Therefore, the exploration of controlling and directing the conformational arrangements is significant in achieving desired nanoarchitectures. Herein, a conformationally flexible precursor 2,4,6-tris(3-bromophenyl)−1,3,5-triazine is employed, and a random phase consisting of C3h and Cs conformers is firstly obtained after deposition of the precursor on Cu(111) at room temperature to 365 K. At low coverage (0.01 ML) selenium doping, we achieve the selectivity of the C3h conformer and improve the nanopore structural homogeneity. The ordered two-dimensional metal organic nanostructure can be fulfilled by selenium doping from room temperature to 365 K. The formation of the conformationally flexible precursor on Cu(111) is explored through the combination of high-resolution scanning tunneling microscopy and non-contact atomic force microscopy. The regulation of energy diagrams in the absence or presence of the Se atom is revealed by density functional theory calculations. These results can enrich the on-surface synthesis toolbox of conformationally flexible precursors, for the design of complex nanoarchitectures, and for future development of engineered nanomaterials. |
| 141. |  | Qing-Xia Chen, Yu-Yang Lu, Yang Yang, Li-Ge Chang, Yi Li, Yuan Yang, Zhen He, Jian-Wei Liu, Yong Ni, Shu-Hong Yu Stress-induced ordering evolution of 1D segmented heteronanostructures and their chemical post-transformations In: Nature Communications, vol. 15, no. 3208, 2024, (The formation mechanisms for periodic heterostructures are still poorly understood. Here, the authors propose a versatile approach to synthesize one-dimensional segmented heterostructures and reveal a stress-induced ordering mechanism through phase-field simulations.
). @article{nokey,
title = {Stress-induced ordering evolution of 1D segmented heteronanostructures and their chemical post-transformations},
author = {Qing-Xia Chen and Yu-Yang Lu and Yang Yang and Li-Ge Chang and Yi Li and Yuan Yang and Zhen He and Jian-Wei Liu and Yong Ni and Shu-Hong Yu },
url = {https://www.nature.com/articles/s41467-024-47446-7.pdf},
doi = {10.1038/s41467-024-47446-7},
year = {2024},
date = {2024-04-13},
urldate = {2024-04-13},
journal = {Nature Communications},
volume = {15},
number = {3208},
abstract = {Investigations of one-dimensional segmented heteronanostructures (1D-SHs) have recently attracted much attention due to their potentials for applications resulting from their structure and synergistic effects between compositions and interfaces. Unfortunately, developing a simple, versatile and controlled synthetic method to fabricate 1D-SHs is still a challenge. Here we demonstrate a stress-induced axial ordering mechanism to describe the synthesis of 1D-SHs by a general under-stoichiometric reaction strategy. Using the continuum phase-field simulations, we elaborate a three-stage evolution process of the regular segment alternations. This strategy, accompanied by easy chemical post-transformations, enables to synthesize 25 1D-SHs, including 17 nanowire-nanowire and 8 nanowire-nanotube nanostructures with 13 elements (Ag, Te, Cu, Pt, Pb, Cd, Sb, Se, Bi, Rh, Ir, Ru, Zn) involved. This ordering evolution-driven synthesis will help to investigate the ordering reconstruction and potential applications of 1D-SHs.},
note = {The formation mechanisms for periodic heterostructures are still poorly understood. Here, the authors propose a versatile approach to synthesize one-dimensional segmented heterostructures and reveal a stress-induced ordering mechanism through phase-field simulations.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Investigations of one-dimensional segmented heteronanostructures (1D-SHs) have recently attracted much attention due to their potentials for applications resulting from their structure and synergistic effects between compositions and interfaces. Unfortunately, developing a simple, versatile and controlled synthetic method to fabricate 1D-SHs is still a challenge. Here we demonstrate a stress-induced axial ordering mechanism to describe the synthesis of 1D-SHs by a general under-stoichiometric reaction strategy. Using the continuum phase-field simulations, we elaborate a three-stage evolution process of the regular segment alternations. This strategy, accompanied by easy chemical post-transformations, enables to synthesize 25 1D-SHs, including 17 nanowire-nanowire and 8 nanowire-nanotube nanostructures with 13 elements (Ag, Te, Cu, Pt, Pb, Cd, Sb, Se, Bi, Rh, Ir, Ru, Zn) involved. This ordering evolution-driven synthesis will help to investigate the ordering reconstruction and potential applications of 1D-SHs. |
| 140. | 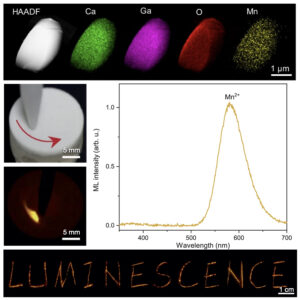 | Yiqian Tang, Yiyu Cai, Kunpeng Dou, Jianqing Chang, Wei Li, Shanshan Wang, Mingzi Sun, Bolong Huang, Xiaofeng Liu, Jianrong Qiu, Lei Zhou, Mingmei Wu, Jun-Cheng Zhang Dynamic multicolor emissions of multimodal phosphors by Mn2+ trace doping in self-activated CaGa4O7 In: Nature Communications, vol. 15, no. 3209, 2024, (Achieving dynamic multimodal luminescence in a single material is promising but challenging. Here, the authors engineer a phosphor with dynamic multicolor luminescence and photo-thermomechanically responsive emissions by adding Mn2+ to a self-activated CaGa4O7 host.). @article{nokey,
title = {Dynamic multicolor emissions of multimodal phosphors by Mn2+ trace doping in self-activated CaGa4O7},
author = {Yiqian Tang and Yiyu Cai and Kunpeng Dou and Jianqing Chang and Wei Li and Shanshan Wang and Mingzi Sun and Bolong Huang and Xiaofeng Liu and Jianrong Qiu and Lei Zhou and Mingmei Wu and Jun-Cheng Zhang },
url = {https://www.nature.com/articles/s41467-024-47431-0.pdf},
doi = {10.1038/s41467-024-47431-0},
year = {2024},
date = {2024-04-13},
urldate = {2024-04-13},
journal = {Nature Communications},
volume = {15},
number = {3209},
abstract = {The manipulation of excitation modes and resultant emission colors in luminescent materials holds pivotal importance for encrypting information in anti-counterfeiting applications. Despite considerable achievements in multimodal and multicolor luminescent materials, existing options generally suffer from static monocolor emission under fixed external stimulation, rendering them vulnerability to replication. Achieving dynamic multimodal luminescence within a single material presents a promising yet challenging solution. Here, we report the development of a phosphor exhibiting dynamic multicolor photoluminescence (PL) and photo-thermo-mechanically responsive multimodal emissions through the incorporation of trace Mn2+ ions into a self-activated CaGa4O7 host. The resulting phosphor offers adjustable emission-color changing rates, controllable via re-excitation intervals and photoexcitation powers. Additionally, it demonstrates temperature-induced color reversal and anti-thermal-quenched emission, alongside reproducible elastic mechanoluminescence (ML) characterized by high mechanical durability. Theoretical calculations elucidate electron transfer pathways dominated by intrinsic interstitial defects and vacancies for dynamic multicolor emission. Mn2+ dopants serve a dual role in stabilizing nearby defects and introducing additional defect levels, enabling flexible multi-responsive luminescence. This developed phosphor facilitates evolutionary color/pattern displays in both temporal and spatial dimensions using readily available tools, offering significant promise for dynamic anticounterfeiting displays and multimode sensing applications.},
note = {Achieving dynamic multimodal luminescence in a single material is promising but challenging. Here, the authors engineer a phosphor with dynamic multicolor luminescence and photo-thermomechanically responsive emissions by adding Mn2+ to a self-activated CaGa4O7 host.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The manipulation of excitation modes and resultant emission colors in luminescent materials holds pivotal importance for encrypting information in anti-counterfeiting applications. Despite considerable achievements in multimodal and multicolor luminescent materials, existing options generally suffer from static monocolor emission under fixed external stimulation, rendering them vulnerability to replication. Achieving dynamic multimodal luminescence within a single material presents a promising yet challenging solution. Here, we report the development of a phosphor exhibiting dynamic multicolor photoluminescence (PL) and photo-thermo-mechanically responsive multimodal emissions through the incorporation of trace Mn2+ ions into a self-activated CaGa4O7 host. The resulting phosphor offers adjustable emission-color changing rates, controllable via re-excitation intervals and photoexcitation powers. Additionally, it demonstrates temperature-induced color reversal and anti-thermal-quenched emission, alongside reproducible elastic mechanoluminescence (ML) characterized by high mechanical durability. Theoretical calculations elucidate electron transfer pathways dominated by intrinsic interstitial defects and vacancies for dynamic multicolor emission. Mn2+ dopants serve a dual role in stabilizing nearby defects and introducing additional defect levels, enabling flexible multi-responsive luminescence. This developed phosphor facilitates evolutionary color/pattern displays in both temporal and spatial dimensions using readily available tools, offering significant promise for dynamic anticounterfeiting displays and multimode sensing applications. |
| 139. | 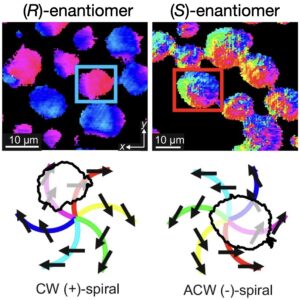 | Alexander P. Fellows, Ben John, Martin Wolf, Martin Thämer Spiral packing and chiral selectivity in model membranes probed by phase-resolved sum-frequency generation microscopy In: Nature Communications, vol. 15, no. 3161, 2024, (The properties of lipid membranes are intimately controlled by their complex heterogeneous structure. Here, the authors use phase-resolved sum-frequency generation microscopy to fully determine the hierarchical lipid packing from the molecular to the mesoscopic scale.). @article{nokey,
title = {Spiral packing and chiral selectivity in model membranes probed by phase-resolved sum-frequency generation microscopy},
author = {Alexander P. Fellows and Ben John and Martin Wolf and Martin Thämer },
url = {https://www.nature.com/articles/s41467-024-47573-1.pdf},
doi = {10.1038/s41467-024-47573-1},
year = {2024},
date = {2024-04-11},
urldate = {2024-04-11},
journal = {Nature Communications},
volume = {15},
number = {3161},
abstract = {Since the lipid raft model was developed at the end of the last century, it became clear that the specific molecular arrangements of phospholipid assemblies within a membrane have profound implications in a vast range of physiological functions. Studies of such condensed lipid islands in model systems using fluorescence and Brewster angle microscopies have shown a wide range of sizes and morphologies, with suggestions of substantial in-plane molecular anisotropy and mesoscopic structural chirality. Whilst these variations can significantly alter many membrane properties including its fluidity, permeability and molecular recognition, the details of the in-plane molecular orientations underlying these traits remain largely unknown. Here, we use phase-resolved sum-frequency generation microscopy on model membranes of mixed chirality phospholipid monolayers to fully determine the three-dimensional molecular structure of the constituent micron-scale condensed domains. We find that the domains possess curved molecular directionality with spiralling mesoscopic packing, where both the molecular and spiral turning directions depend on the lipid chirality, but form structures clearly deviating from mirror symmetry for different enantiomeric mixtures. This demonstrates strong enantioselectivity in the domain growth process and indicates fundamental thermodynamic differences between homo- and heterochiral membranes, which may be relevant in the evolution of homochirality in all living organisms.},
note = {The properties of lipid membranes are intimately controlled by their complex heterogeneous structure. Here, the authors use phase-resolved sum-frequency generation microscopy to fully determine the hierarchical lipid packing from the molecular to the mesoscopic scale.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Since the lipid raft model was developed at the end of the last century, it became clear that the specific molecular arrangements of phospholipid assemblies within a membrane have profound implications in a vast range of physiological functions. Studies of such condensed lipid islands in model systems using fluorescence and Brewster angle microscopies have shown a wide range of sizes and morphologies, with suggestions of substantial in-plane molecular anisotropy and mesoscopic structural chirality. Whilst these variations can significantly alter many membrane properties including its fluidity, permeability and molecular recognition, the details of the in-plane molecular orientations underlying these traits remain largely unknown. Here, we use phase-resolved sum-frequency generation microscopy on model membranes of mixed chirality phospholipid monolayers to fully determine the three-dimensional molecular structure of the constituent micron-scale condensed domains. We find that the domains possess curved molecular directionality with spiralling mesoscopic packing, where both the molecular and spiral turning directions depend on the lipid chirality, but form structures clearly deviating from mirror symmetry for different enantiomeric mixtures. This demonstrates strong enantioselectivity in the domain growth process and indicates fundamental thermodynamic differences between homo- and heterochiral membranes, which may be relevant in the evolution of homochirality in all living organisms. |
| 138. |  | Guoying Bai, Haiyan Zhang, Dong Gao, Houguo Fei, Cunlan Guo, Mingxia Ren, Yufeng Liu Controlled condensation by liquid contact-induced adaptations of molecular conformations in self-assembled monolayers In: Nature Communications, vol. 2024, no. 3132, 2024, (Surface condensation is predetermined and is typically adjusted by chemical or topographical surface modification. Here, the authors report on a strategy to control the surface condensation behavior by adjusting molecular conformations in self-assembled monolayers.
). @article{nokey,
title = {Controlled condensation by liquid contact-induced adaptations of molecular conformations in self-assembled monolayers},
author = {Guoying Bai and Haiyan Zhang and Dong Gao and Houguo Fei and Cunlan Guo and Mingxia Ren and Yufeng Liu },
url = {https://www.nature.com/articles/s41467-024-47507-x.pdf},
doi = {10.1038/s41467-024-47507-x},
year = {2024},
date = {2024-04-11},
urldate = {2024-04-11},
journal = {Nature Communications},
volume = {2024},
number = {3132},
abstract = {Surface condensation control strategies are crucial but commonly require relatively tedious, time-consuming, and expensive techniques for surface-chemical and topographical engineering. Here we report a strategy to alter surface condensation behavior without resorting to any molecule-type or topographical transmutations. After ultrafast contact of liquids with and removal from surfaces, the condensation rate and density of water droplets on the surfaces decrease, the extent of which is positively correlated with the polarity of the liquid and the duration of contact. The liquid contact-induced condensation rate/density decrease (LCICD) can be attributed to the decrease of nucleation site density resulted from the liquid contact-induced adaption of surface molecular conformation. Based on this, we find that LCICD is applicable to various surfaces, on condition that there are flexible segments capable of shielding at least part of nucleation sites through changing the conformation under liquid contact induction. Leveraging the LCICD effect, we achieve erasable information storage on diverse substrates. Furthermore, our strategy holds promise for controlling condensation of other substances since LCICD is not specific to the water condensation process.},
note = {Surface condensation is predetermined and is typically adjusted by chemical or topographical surface modification. Here, the authors report on a strategy to control the surface condensation behavior by adjusting molecular conformations in self-assembled monolayers.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Surface condensation control strategies are crucial but commonly require relatively tedious, time-consuming, and expensive techniques for surface-chemical and topographical engineering. Here we report a strategy to alter surface condensation behavior without resorting to any molecule-type or topographical transmutations. After ultrafast contact of liquids with and removal from surfaces, the condensation rate and density of water droplets on the surfaces decrease, the extent of which is positively correlated with the polarity of the liquid and the duration of contact. The liquid contact-induced condensation rate/density decrease (LCICD) can be attributed to the decrease of nucleation site density resulted from the liquid contact-induced adaption of surface molecular conformation. Based on this, we find that LCICD is applicable to various surfaces, on condition that there are flexible segments capable of shielding at least part of nucleation sites through changing the conformation under liquid contact induction. Leveraging the LCICD effect, we achieve erasable information storage on diverse substrates. Furthermore, our strategy holds promise for controlling condensation of other substances since LCICD is not specific to the water condensation process. |
| 137. | 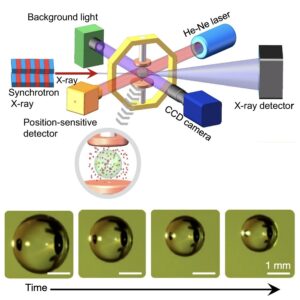 | Yong Chan Cho, Sooheyong Lee, Lei Wang, Yun-Hee Lee, Seongheun Kim, Hyun-Hwi Lee, John Jonghyun Lee, Geun Woo Lee Impact of molecular symmetry on crystallization pathways in highly supersaturated KH2PO4 solutions In: Nature Communications, vol. 15, no. 3117, 2024, (The molecular symmetry of solute structure in aqueous solutions is a key clue to understand Ostwald’s step rule. Here, the authors show that molecular symmetry and its structural evolution can govern the crystallization pathways in aqueous solutions.). @article{nokey,
title = {Impact of molecular symmetry on crystallization pathways in highly supersaturated KH2PO4 solutions},
author = {Yong Chan Cho and Sooheyong Lee and Lei Wang and Yun-Hee Lee and Seongheun Kim and Hyun-Hwi Lee and John Jonghyun Lee and Geun Woo Lee },
url = {https://www.nature.com/articles/s41467-024-47503-1.pdf},
doi = {10.1038/s41467-024-47503-1},
year = {2024},
date = {2024-04-10},
urldate = {2024-04-10},
journal = {Nature Communications},
volume = {15},
number = {3117},
abstract = {Solute structure and its evolution in supersaturated aqueous solutions are key clues to understand Ostwald’s step rule. Here, we measure the structural evolution of solute molecules in highly supersaturated solutions of KH2PO4 (KDP) and NH4H2PO4 (ADP) using a combination of electrostatic levitation and synchrotron X-ray scattering. The measurement reveals the existence of a solution-solution transition in KDP solution, caused by changing molecular symmetries and structural evolution of the solution with supersaturation. Moreover, we find that the molecular symmetry of H2PO4- impacts on phase selection. These findings manifest that molecular symmetry and its structural evolution can govern the crystallization pathways in aqueous solutions, explaining the microscopic origin of Ostwald’s step rule.},
note = {The molecular symmetry of solute structure in aqueous solutions is a key clue to understand Ostwald’s step rule. Here, the authors show that molecular symmetry and its structural evolution can govern the crystallization pathways in aqueous solutions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Solute structure and its evolution in supersaturated aqueous solutions are key clues to understand Ostwald’s step rule. Here, we measure the structural evolution of solute molecules in highly supersaturated solutions of KH2PO4 (KDP) and NH4H2PO4 (ADP) using a combination of electrostatic levitation and synchrotron X-ray scattering. The measurement reveals the existence of a solution-solution transition in KDP solution, caused by changing molecular symmetries and structural evolution of the solution with supersaturation. Moreover, we find that the molecular symmetry of H2PO4- impacts on phase selection. These findings manifest that molecular symmetry and its structural evolution can govern the crystallization pathways in aqueous solutions, explaining the microscopic origin of Ostwald’s step rule. |
| 136. |  | Soosang Chae, Won Jin Choi, Lisa Julia Nebel, Chang Hee Cho, Quinn A. Besford, André Knapp, Pavlo Makushko, Yevhen Zabila, Oleksandr Pylypovskyi, Min Woo Jeong, Stanislav Avdoshenko, Oliver Sander, Denys Makarov, Yoon Jang Chung, Andreas Fery, Jin Young Oh, Tae Il Lee Kinetically controlled metal-elastomer nanophases for environmentally resilient stretchable electronics In: Nature Communications, vol. 15, no. 3071, 2024, (Metal-elastomer nanophases are critical for stretchable electronics but face mixing challenges. This study introduces a kinetic method for precise mixing, yielding gyrified nanophases with improved durability and strain-invariant conductivity, which holds promise for resilient stretchable devices.). @article{nokey,
title = {Kinetically controlled metal-elastomer nanophases for environmentally resilient stretchable electronics},
author = {Soosang Chae and Won Jin Choi and Lisa Julia Nebel and Chang Hee Cho and Quinn A. Besford and André Knapp and Pavlo Makushko and Yevhen Zabila and Oleksandr Pylypovskyi and Min Woo Jeong and Stanislav Avdoshenko and Oliver Sander and Denys Makarov and Yoon Jang Chung and Andreas Fery and Jin Young Oh and Tae Il Lee },
url = {https://www.nature.com/articles/s41467-024-47223-6.pdf},
doi = {10.1038/s41467-024-47223-6},
year = {2024},
date = {2024-04-09},
urldate = {2024-04-09},
journal = {Nature Communications},
volume = {15},
number = {3071},
abstract = {Nanophase mixtures, leveraging the complementary strengths of each component, are vital for composites to overcome limitations posed by single elemental materials. Among these, metal-elastomer nanophases are particularly important, holding various practical applications for stretchable electronics. However, the methodology and understanding of nanophase mixing metals and elastomers are limited due to difficulties in blending caused by thermodynamic incompatibility. Here, we present a controlled method using kinetics to mix metal atoms with elastomeric chains on the nanoscale. We find that the chain migration flux and metal deposition rate are key factors, allowing the formation of reticular nanophases when kinetically in-phase. Moreover, we observe spontaneous structural evolution, resulting in gyrified structures akin to the human brain. The hybridized gyrified reticular nanophases exhibit strain-invariant metallic electrical conductivity up to 156% areal strain, unparalleled durability in organic solvents and aqueous environments with pH 2–13, and high mechanical robustness, a prerequisite for environmentally resilient devices.},
note = {Metal-elastomer nanophases are critical for stretchable electronics but face mixing challenges. This study introduces a kinetic method for precise mixing, yielding gyrified nanophases with improved durability and strain-invariant conductivity, which holds promise for resilient stretchable devices.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanophase mixtures, leveraging the complementary strengths of each component, are vital for composites to overcome limitations posed by single elemental materials. Among these, metal-elastomer nanophases are particularly important, holding various practical applications for stretchable electronics. However, the methodology and understanding of nanophase mixing metals and elastomers are limited due to difficulties in blending caused by thermodynamic incompatibility. Here, we present a controlled method using kinetics to mix metal atoms with elastomeric chains on the nanoscale. We find that the chain migration flux and metal deposition rate are key factors, allowing the formation of reticular nanophases when kinetically in-phase. Moreover, we observe spontaneous structural evolution, resulting in gyrified structures akin to the human brain. The hybridized gyrified reticular nanophases exhibit strain-invariant metallic electrical conductivity up to 156% areal strain, unparalleled durability in organic solvents and aqueous environments with pH 2–13, and high mechanical robustness, a prerequisite for environmentally resilient devices. |
| 135. |  | Junbo Wang, Kaifeng Niu, Huaming Zhu, Chaojie Xu, Chuan Deng, Wenchao Zhao, Peipei Huang, Haiping Lin, Dengyuan Li, Johanna Rosen, Peinian Liu, Francesco Allegretti, Johannes V. Barth, Biao Yang, Jonas Björk, Qing Li, Lifeng Chi Universal inter-molecular radical transfer reactions on metal surfaces In: Nature Communications, vol. 15, no. 3030, 2024, (Radicals are expected to be inactive on metal surfaces. Here the authors describe general intermolecular radical transfer reactions on Ag and Cu surfaces and confirm the reaction mechanism by extensive control experiments.
). @article{Wang2024,
title = {Universal inter-molecular radical transfer reactions on metal surfaces},
author = {Junbo Wang and Kaifeng Niu and Huaming Zhu and Chaojie Xu and Chuan Deng and Wenchao Zhao and Peipei Huang and Haiping Lin and Dengyuan Li and Johanna Rosen and Peinian Liu and Francesco Allegretti and Johannes V. Barth and Biao Yang and Jonas Björk and Qing Li and Lifeng Chi },
url = {https://www.nature.com/articles/s41467-024-47252-1.pdf},
doi = {10.1038/s41467-024-47252-1},
year = {2024},
date = {2024-04-08},
urldate = {2024-04-08},
journal = {Nature Communications},
volume = {15},
number = {3030},
abstract = {On-surface synthesis provides tools to prepare low-dimensional supramolecular structures. Traditionally, reactive radicals are a class of single-electron species, serving as exceptional electron-withdrawing groups. On metal surfaces, however, such species are affected by conduction band screening effects that may even quench their unpaired electron characteristics. As a result, radicals are expected to be less active, and reactions catalyzed by surface-stabilized radicals are rarely reported. Herein, we describe a class of inter-molecular radical transfer reactions on metal surfaces. With the assistance of aryl halide precursors, the coupling of terminal alkynes is steered from non-dehydrogenated to dehydrogenated products, resulting in alkynyl-Ag-alkynyl bonds. Dehalogenated molecules are fully passivated by detached hydrogen atoms. The reaction mechanism is unraveled by various surface-sensitive technologies and density functional theory calculations. Moreover, we reveal the universality of this mechanism on metal surfaces. Our studies enrich the on-surface synthesis toolbox and develop a pathway for producing low-dimensional organic materials.},
note = {Radicals are expected to be inactive on metal surfaces. Here the authors describe general intermolecular radical transfer reactions on Ag and Cu surfaces and confirm the reaction mechanism by extensive control experiments.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
On-surface synthesis provides tools to prepare low-dimensional supramolecular structures. Traditionally, reactive radicals are a class of single-electron species, serving as exceptional electron-withdrawing groups. On metal surfaces, however, such species are affected by conduction band screening effects that may even quench their unpaired electron characteristics. As a result, radicals are expected to be less active, and reactions catalyzed by surface-stabilized radicals are rarely reported. Herein, we describe a class of inter-molecular radical transfer reactions on metal surfaces. With the assistance of aryl halide precursors, the coupling of terminal alkynes is steered from non-dehydrogenated to dehydrogenated products, resulting in alkynyl-Ag-alkynyl bonds. Dehalogenated molecules are fully passivated by detached hydrogen atoms. The reaction mechanism is unraveled by various surface-sensitive technologies and density functional theory calculations. Moreover, we reveal the universality of this mechanism on metal surfaces. Our studies enrich the on-surface synthesis toolbox and develop a pathway for producing low-dimensional organic materials. |
| 134. |  | Xiaoli Tian, Fu Li, Zhenyuan Tang, Song Wang, Kangkang Weng, Dan Liu, Shaoyong Lu, Wangyu Liu, Zhong Fu, Wenjun Li, Hengwei Qiu, Min Tu, Hao Zhang, Jinghong Li Crosslinking-induced patterning of MOFs by direct photo- and electron-beam lithography In: Nature Communications, vol. 15, no. 2920, 2024, (Precise and scalable patterning is essential for the use of metal-organic frameworks (MOFs) in solid-state electronics and photonics. Here, the authors report on resistance-free, direct photo- and electron-beam lithography of MOF films using crosslinking chemistry.). @article{nokey,
title = {Crosslinking-induced patterning of MOFs by direct photo- and electron-beam lithography},
author = {Xiaoli Tian and Fu Li and Zhenyuan Tang and Song Wang and Kangkang Weng and Dan Liu and Shaoyong Lu and Wangyu Liu and Zhong Fu and Wenjun Li and Hengwei Qiu and Min Tu and Hao Zhang and Jinghong Li },
url = {https://www.nature.com/articles/s41467-024-47293-6.pdf},
doi = {10.1038/s41467-024-47293-6},
year = {2024},
date = {2024-04-04},
urldate = {2024-04-04},
journal = {Nature Communications},
volume = {15},
number = {2920},
abstract = {Metal-organic frameworks (MOFs) with diverse chemistry, structures, and properties have emerged as appealing materials for miniaturized solid-state devices. The incorporation of MOF films in these devices, such as the integrated microelectronics and nanophotonics, requires robust patterning methods. However, existing MOF patterning methods suffer from some combinations of limited material adaptability, compromised patterning resolution and scalability, and degraded properties. Here we report a universal, crosslinking-induced patterning approach for various MOFs, termed as CLIP-MOF. Via resist-free, direct photo- and electron-beam (e-beam) lithography, the ligand crosslinking chemistry leads to drastically reduced solubility of colloidal MOFs, permitting selective removal of unexposed MOF films with developer solvents. This enables scalable, micro-/nanoscale (≈70 nm resolution), and multimaterial patterning of MOFs on large-area, rigid or flexible substrates. Patterned MOF films preserve their crystallinity, porosity, and other properties tailored for targeted applications, such as diffractive gas sensors and electrochromic pixels. The combined features of CLIP-MOF create more possibilities in the system-level integration of MOFs in various electronic, photonic, and biomedical devices.},
note = {Precise and scalable patterning is essential for the use of metal-organic frameworks (MOFs) in solid-state electronics and photonics. Here, the authors report on resistance-free, direct photo- and electron-beam lithography of MOF films using crosslinking chemistry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Metal-organic frameworks (MOFs) with diverse chemistry, structures, and properties have emerged as appealing materials for miniaturized solid-state devices. The incorporation of MOF films in these devices, such as the integrated microelectronics and nanophotonics, requires robust patterning methods. However, existing MOF patterning methods suffer from some combinations of limited material adaptability, compromised patterning resolution and scalability, and degraded properties. Here we report a universal, crosslinking-induced patterning approach for various MOFs, termed as CLIP-MOF. Via resist-free, direct photo- and electron-beam (e-beam) lithography, the ligand crosslinking chemistry leads to drastically reduced solubility of colloidal MOFs, permitting selective removal of unexposed MOF films with developer solvents. This enables scalable, micro-/nanoscale (≈70 nm resolution), and multimaterial patterning of MOFs on large-area, rigid or flexible substrates. Patterned MOF films preserve their crystallinity, porosity, and other properties tailored for targeted applications, such as diffractive gas sensors and electrochromic pixels. The combined features of CLIP-MOF create more possibilities in the system-level integration of MOFs in various electronic, photonic, and biomedical devices. |
| 133. |  | David Doan, John Kulikowski, X. Wendy Gu Direct observation of phase transitions in truncated tetrahedral microparticles under quasi-2D confinement In: Nature Communications, vol. 15, no. 1954, 2024, (Boundary conditions can give rise to new types of phases during self-assembly. Here the authors show that tetrahedral particles can form a hexagonal phase on a surface, that can transform into a quasi-diamond phase under a gravitational field.). @article{nokey,
title = {Direct observation of phase transitions in truncated tetrahedral microparticles under quasi-2D confinement},
author = {David Doan and John Kulikowski and X. Wendy Gu },
url = {https://www.nature.com/articles/s41467-024-46230-x.pdf},
doi = {10.1038/s41467-024-46230-x},
year = {2024},
date = {2024-03-25},
urldate = {2024-03-25},
journal = {Nature Communications},
volume = {15},
number = {1954},
abstract = {Colloidal crystals are used to understand fundamentals of atomic rearrangements in condensed matter and build complex metamaterials with unique functionalities. Simulations predict a multitude of self-assembled crystal structures from anisotropic colloids, but these shapes have been challenging to fabricate. Here, we use two-photon lithography to fabricate Archimedean truncated tetrahedrons and self-assemble them under quasi-2D confinement. These particles self-assemble into a hexagonal phase under an in-plane gravitational potential. Under additional gravitational potential, the hexagonal phase transitions into a quasi-diamond two-unit basis. In-situ imaging reveal this phase transition is initiated by an out-of-plane rotation of a particle at a crystalline defect and causes a chain reaction of neighboring particle rotations. Our results provide a framework of studying different structures from hard-particle self-assembly and demonstrates the ability to use confinement to induce unusual phases.},
note = {Boundary conditions can give rise to new types of phases during self-assembly. Here the authors show that tetrahedral particles can form a hexagonal phase on a surface, that can transform into a quasi-diamond phase under a gravitational field.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Colloidal crystals are used to understand fundamentals of atomic rearrangements in condensed matter and build complex metamaterials with unique functionalities. Simulations predict a multitude of self-assembled crystal structures from anisotropic colloids, but these shapes have been challenging to fabricate. Here, we use two-photon lithography to fabricate Archimedean truncated tetrahedrons and self-assemble them under quasi-2D confinement. These particles self-assemble into a hexagonal phase under an in-plane gravitational potential. Under additional gravitational potential, the hexagonal phase transitions into a quasi-diamond two-unit basis. In-situ imaging reveal this phase transition is initiated by an out-of-plane rotation of a particle at a crystalline defect and causes a chain reaction of neighboring particle rotations. Our results provide a framework of studying different structures from hard-particle self-assembly and demonstrates the ability to use confinement to induce unusual phases. |
| 132. |  | Emily Xi Tan, Shi Xuan Leong, Wei An Liew, In Yee Phang, Jie Ying Ng, Nguan Soon Tan, Yie Hou Lee, Xing Yi Ling
Forward-predictive SERS-based chemical taxonomy for untargeted structural elucidation of epimeric cerebrosides In: Nature Communications, vol. 15, no. 2582, 2024, (SERS-based chemical taxonomy is attractive for practical SERS applications where the identity and concentration of analytes are unknown. Here, the authors demonstrate a machine learning framework for classifying “unknown” molecules that lie outside the boundaries of the models used.). @article{nokey,
title = {Forward-predictive SERS-based chemical taxonomy for untargeted structural elucidation of epimeric cerebrosides},
author = {Emily Xi Tan and Shi Xuan Leong and Wei An Liew and In Yee Phang and Jie Ying Ng and Nguan Soon Tan and Yie Hou Lee and Xing Yi Ling
},
url = {https://www.nature.com/articles/s41467-024-46838-z.pdf},
doi = {10.1038/s41467-024-46838-z},
year = {2024},
date = {2024-03-22},
urldate = {2024-03-22},
journal = {Nature Communications},
volume = {15},
number = {2582},
abstract = {Achieving untargeted chemical identification, isomeric differentiation, and quantification is critical to most scientific and technological problems but remains challenging. Here, we demonstrate an integrated SERS-based chemical taxonomy machine learning framework for untargeted structural elucidation of 11 epimeric cerebrosides, attaining >90% accuracy and robust single epimer and multiplex quantification with <10% errors. First, we utilize 4-mercaptophenylboronic acid to selectively capture the epimers at molecular sites of isomerism to form epimer-specific SERS fingerprints. Corroborating with in-silico experiments, we establish five spectral features, each corresponding to a structural characteristic: (1) presence/absence of epimers, (2) monosaccharide/cerebroside, (3) saturated/unsaturated cerebroside, (4) glucosyl/galactosyl, and (5) GlcCer or GalCer’s carbon chain lengths. Leveraging these insights, we create a fully generalizable framework to identify and quantify cerebrosides at concentrations between 10−4 to 10−10 M and achieve multiplex quantification of binary mixtures containing biomarkers GlcCer24:1, and GalCer24:1 using their untrained spectra in the models},
note = {SERS-based chemical taxonomy is attractive for practical SERS applications where the identity and concentration of analytes are unknown. Here, the authors demonstrate a machine learning framework for classifying “unknown” molecules that lie outside the boundaries of the models used.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Achieving untargeted chemical identification, isomeric differentiation, and quantification is critical to most scientific and technological problems but remains challenging. Here, we demonstrate an integrated SERS-based chemical taxonomy machine learning framework for untargeted structural elucidation of 11 epimeric cerebrosides, attaining >90% accuracy and robust single epimer and multiplex quantification with <10% errors. First, we utilize 4-mercaptophenylboronic acid to selectively capture the epimers at molecular sites of isomerism to form epimer-specific SERS fingerprints. Corroborating with in-silico experiments, we establish five spectral features, each corresponding to a structural characteristic: (1) presence/absence of epimers, (2) monosaccharide/cerebroside, (3) saturated/unsaturated cerebroside, (4) glucosyl/galactosyl, and (5) GlcCer or GalCer’s carbon chain lengths. Leveraging these insights, we create a fully generalizable framework to identify and quantify cerebrosides at concentrations between 10−4 to 10−10 M and achieve multiplex quantification of binary mixtures containing biomarkers GlcCer24:1, and GalCer24:1 using their untrained spectra in the models |
| 131. |  | Stefan Merkens, Christopher Tollan, Giuseppe De Salvo, Katarzyna Bejtka, Marco Fontana, Angelica Chiodoni, Joscha Kruse, Maiara Aime Iriarte-Alonso, Marek Grzelczak, Andreas Seifert, Andrey Chuvilin Toward sub-second solution exchange dynamics in flow reactors for liquid-phase transmission electron microscopy In: Nature Communications, vol. 15, no. 2522, 2024, (In liquid-phase TEM, microfluidic reactors are used to monitor nanoscale (electro)chemical dynamics in liquid environments. Here, the authors develop a reactor design with accelerated mass transport, facilitating quantitative in-situ and in-operando studies.). @article{nokey,
title = {Toward sub-second solution exchange dynamics in flow reactors for liquid-phase transmission electron microscopy},
author = {Stefan Merkens and Christopher Tollan and Giuseppe De Salvo and Katarzyna Bejtka and Marco Fontana and Angelica Chiodoni and Joscha Kruse and Maiara Aime Iriarte-Alonso and Marek Grzelczak and Andreas Seifert and Andrey Chuvilin },
url = {https://www.nature.com/articles/s41467-024-46842-3.pdf},
doi = {10.1038/s41467-024-46842-3},
year = {2024},
date = {2024-03-21},
urldate = {2024-03-21},
journal = {Nature Communications},
volume = {15},
number = {2522},
abstract = {Liquid-phase transmission electron microscopy is a burgeoning experimental technique for monitoring nanoscale dynamics in a liquid environment, increasingly employing microfluidic reactors to control the composition of the sample solution. Current challenges comprise fast mass transport dynamics inside the central nanochannel of the liquid cell, typically flow cells, and reliable fixation of the specimen in the limited imaging area. In this work, we present a liquid cell concept – the diffusion cell – that satisfies these seemingly contradictory requirements by providing additional on-chip bypasses to allow high convective transport around the nanochannel in which diffusive transport predominates. Diffusion cell prototypes are developed using numerical mass transport models and fabricated on the basis of existing two-chip setups. Important hydrodynamic parameters, i.e., the total flow resistance, the flow velocity in the imaging area, and the time constants of mixing, are improved by 2-3 orders of magnitude compared to existing setups. The solution replacement dynamics achieved within seconds already match the mixing timescales of many ex-situ scenarios, and further improvements are possible. Diffusion cells can be easily integrated into existing liquid-phase transmission electron microscopy workflows, provide correlation of results with ex-situ experiments, and can create additional research directions addressing fast nanoscale processes.},
note = {In liquid-phase TEM, microfluidic reactors are used to monitor nanoscale (electro)chemical dynamics in liquid environments. Here, the authors develop a reactor design with accelerated mass transport, facilitating quantitative in-situ and in-operando studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Liquid-phase transmission electron microscopy is a burgeoning experimental technique for monitoring nanoscale dynamics in a liquid environment, increasingly employing microfluidic reactors to control the composition of the sample solution. Current challenges comprise fast mass transport dynamics inside the central nanochannel of the liquid cell, typically flow cells, and reliable fixation of the specimen in the limited imaging area. In this work, we present a liquid cell concept – the diffusion cell – that satisfies these seemingly contradictory requirements by providing additional on-chip bypasses to allow high convective transport around the nanochannel in which diffusive transport predominates. Diffusion cell prototypes are developed using numerical mass transport models and fabricated on the basis of existing two-chip setups. Important hydrodynamic parameters, i.e., the total flow resistance, the flow velocity in the imaging area, and the time constants of mixing, are improved by 2-3 orders of magnitude compared to existing setups. The solution replacement dynamics achieved within seconds already match the mixing timescales of many ex-situ scenarios, and further improvements are possible. Diffusion cells can be easily integrated into existing liquid-phase transmission electron microscopy workflows, provide correlation of results with ex-situ experiments, and can create additional research directions addressing fast nanoscale processes. |
| 130. | 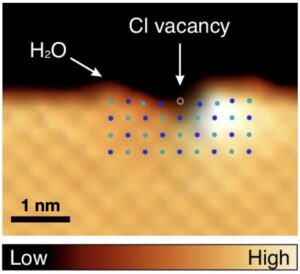 | Huijun Han, Yunjae Park, Yohan Kim, Feng Ding, Hyung-Joon Shin Controlled dissolution of a single ion from a salt interface In: Nature Communications, vol. 15, no. 2401, 2024, (The strong ionic bond in salt is broken by electrostatic interactions with water, but direct observation at the level of a single ion is challenging. Here, the authors have visualized the preferential dissolution of an anion by manipulating a single water molecule.). @article{nokey,
title = {Controlled dissolution of a single ion from a salt interface},
author = {Huijun Han and Yunjae Park and Yohan Kim and Feng Ding and Hyung-Joon Shin },
url = {https://www.nature.com/articles/s41467-024-46704-y.pdf},
doi = {10.1038/s41467-024-46704-y},
year = {2024},
date = {2024-03-16},
urldate = {2024-03-16},
journal = {Nature Communications},
volume = {15},
number = {2401},
abstract = {Interactions between monatomic ions and water molecules are fundamental to understanding the hydration of complex polyatomic ions and ionic process. Among the simplest and well-established ion-related reactions is dissolution of salt in water, which is an endothermic process requiring an increase in entropy. Extensive efforts have been made to date; however, most studies at single-ion level have been limited to theoretical approaches. Here, we demonstrate the salt dissolution process by manipulating a single water molecule at an under-coordinated site of a sodium chloride film. Manipulation of molecule in a controlled manner enables us to understand ion–water interaction as well as dynamics of water molecules at NaCl interfaces, which are responsible for the selective dissolution of anions. The water dipole polarizes the anion in the NaCl ionic crystal, resulting in strong anion–water interaction and weakening of the ionic bonds. Our results provide insights into a simple but important elementary step of the single-ion chemistry, which may be useful in ion-related sciences and technologies.
},
note = {The strong ionic bond in salt is broken by electrostatic interactions with water, but direct observation at the level of a single ion is challenging. Here, the authors have visualized the preferential dissolution of an anion by manipulating a single water molecule.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Interactions between monatomic ions and water molecules are fundamental to understanding the hydration of complex polyatomic ions and ionic process. Among the simplest and well-established ion-related reactions is dissolution of salt in water, which is an endothermic process requiring an increase in entropy. Extensive efforts have been made to date; however, most studies at single-ion level have been limited to theoretical approaches. Here, we demonstrate the salt dissolution process by manipulating a single water molecule at an under-coordinated site of a sodium chloride film. Manipulation of molecule in a controlled manner enables us to understand ion–water interaction as well as dynamics of water molecules at NaCl interfaces, which are responsible for the selective dissolution of anions. The water dipole polarizes the anion in the NaCl ionic crystal, resulting in strong anion–water interaction and weakening of the ionic bonds. Our results provide insights into a simple but important elementary step of the single-ion chemistry, which may be useful in ion-related sciences and technologies.
|
| 129. |  | Rustem Bolat, Jose M. Guevara, Philipp Leinen, Marvin Knol, Hadi H. Arefi, Michael Maiworm, Rolf Findeisen, Ruslan Temirov, Oliver T. Hofmann, Reinhard J. Maurer, F. Stefan Tautz, Christian Wagner Electrostatic potentials of atomic nanostructures at metal surfaces quantified by scanning quantum dot microscopy In: Nature Communications, vol. 15, no. 2259, 2024, (Surface averaging techniques offer only limited access to the electrostatic potentials of nanostructures, which are determined by shape, material, and environment. Here, the authors quantify these potentials for gold and silver adatom chains, explaining the mechanisms of dipole formation.). @article{nokey,
title = {Electrostatic potentials of atomic nanostructures at metal surfaces quantified by scanning quantum dot microscopy},
author = {Rustem Bolat and Jose M. Guevara and Philipp Leinen and Marvin Knol and Hadi H. Arefi and Michael Maiworm and Rolf Findeisen and Ruslan Temirov and Oliver T. Hofmann and Reinhard J. Maurer and F. Stefan Tautz and Christian Wagner },
url = {https://www.nature.com/articles/s41467-024-46423-4.pdf},
doi = {10.1038/s41467-024-46423-4},
year = {2024},
date = {2024-03-13},
urldate = {2024-03-13},
journal = {Nature Communications},
volume = {15},
number = {2259},
abstract = {The discrete and charge-separated nature of matter — electrons and nuclei — results in local electrostatic fields that are ubiquitous in nanoscale structures and relevant in catalysis, nanoelectronics and quantum nanoscience. Surface-averaging techniques provide only limited experimental access to these potentials, which are determined by the shape, material, and environment of the nanostructure. Here, we image the potential over adatoms, chains, and clusters of Ag and Au atoms assembled on Ag(111) and quantify their surface dipole moments. By focusing on the total charge density, these data establish a benchmark for theory. Our density functional theory calculations show a very good agreement with experiment and allow a deeper analysis of the dipole formation mechanisms, their dependence on fundamental atomic properties and on the shape of the nanostructures. We formulate an intuitive picture of the basic mechanisms behind dipole formation, allowing better design choices for future nanoscale systems such as single-atom catalysts.},
note = {Surface averaging techniques offer only limited access to the electrostatic potentials of nanostructures, which are determined by shape, material, and environment. Here, the authors quantify these potentials for gold and silver adatom chains, explaining the mechanisms of dipole formation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The discrete and charge-separated nature of matter — electrons and nuclei — results in local electrostatic fields that are ubiquitous in nanoscale structures and relevant in catalysis, nanoelectronics and quantum nanoscience. Surface-averaging techniques provide only limited experimental access to these potentials, which are determined by the shape, material, and environment of the nanostructure. Here, we image the potential over adatoms, chains, and clusters of Ag and Au atoms assembled on Ag(111) and quantify their surface dipole moments. By focusing on the total charge density, these data establish a benchmark for theory. Our density functional theory calculations show a very good agreement with experiment and allow a deeper analysis of the dipole formation mechanisms, their dependence on fundamental atomic properties and on the shape of the nanostructures. We formulate an intuitive picture of the basic mechanisms behind dipole formation, allowing better design choices for future nanoscale systems such as single-atom catalysts. |
| 128. |  | Sarah May Sibug-Torres, David-Benjamin Grys, Gyeongwon Kang, Marika Niihori, Elle Wyatt, Nicolas Spiesshofer, Ashleigh Ruane, Bart de Nijs, Jeremy J. Baumberg In situ electrochemical regeneration of nanogap hotspots for continuously reusable ultrathin SERS sensors In: Nature Communications, vol. 15, no. 2022, 2024, (SERS is a powerful analytical technique, but achieving reproducibility for continuous analysis a challenge. Here, the authors report a SERS substrate recycling method that enables direct analysis of complex samples without substrate contamination.). @article{nokey,
title = {In situ electrochemical regeneration of nanogap hotspots for continuously reusable ultrathin SERS sensors},
author = {Sarah May Sibug-Torres and David-Benjamin Grys and Gyeongwon Kang and Marika Niihori and Elle Wyatt and Nicolas Spiesshofer and Ashleigh Ruane and Bart de Nijs and Jeremy J. Baumberg },
url = {https://www.nature.com/articles/s41467-024-46097-y.pdf},
doi = {10.1038/s41467-024-46097-y},
year = {2024},
date = {2024-03-06},
urldate = {2024-03-06},
journal = {Nature Communications},
volume = {15},
number = {2022},
abstract = {Surface-enhanced Raman spectroscopy (SERS) harnesses the confinement of light into metallic nanoscale hotspots to achieve highly sensitive label-free molecular detection that can be applied for a broad range of sensing applications. However, challenges related to irreversible analyte binding, substrate reproducibility, fouling, and degradation hinder its widespread adoption. Here we show how in-situ electrochemical regeneration can rapidly and precisely reform the nanogap hotspots to enable the continuous reuse of gold nanoparticle monolayers for SERS. Applying an oxidising potential of +1.5 V (vs Ag/AgCl) for 10 s strips a broad range of adsorbates from the nanogaps and forms a metastable oxide layer of few-monolayer thickness. Subsequent application of a reducing potential of −0.80 V for 5 s in the presence of a nanogap-stabilising molecular scaffold, cucurbit[5]uril, reproducibly regenerates the optimal plasmonic properties with SERS enhancement factors ≈106. The regeneration of the nanogap hotspots allows these SERS substrates to be reused over multiple cycles, demonstrating ≈5% relative standard deviation over at least 30 cycles of analyte detection and regeneration. Such continuous and reliable SERS-based flow analysis accesses diverse applications from environmental monitoring to medical diagnostics.},
note = {SERS is a powerful analytical technique, but achieving reproducibility for continuous analysis a challenge. Here, the authors report a SERS substrate recycling method that enables direct analysis of complex samples without substrate contamination.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Surface-enhanced Raman spectroscopy (SERS) harnesses the confinement of light into metallic nanoscale hotspots to achieve highly sensitive label-free molecular detection that can be applied for a broad range of sensing applications. However, challenges related to irreversible analyte binding, substrate reproducibility, fouling, and degradation hinder its widespread adoption. Here we show how in-situ electrochemical regeneration can rapidly and precisely reform the nanogap hotspots to enable the continuous reuse of gold nanoparticle monolayers for SERS. Applying an oxidising potential of +1.5 V (vs Ag/AgCl) for 10 s strips a broad range of adsorbates from the nanogaps and forms a metastable oxide layer of few-monolayer thickness. Subsequent application of a reducing potential of −0.80 V for 5 s in the presence of a nanogap-stabilising molecular scaffold, cucurbit[5]uril, reproducibly regenerates the optimal plasmonic properties with SERS enhancement factors ≈106. The regeneration of the nanogap hotspots allows these SERS substrates to be reused over multiple cycles, demonstrating ≈5% relative standard deviation over at least 30 cycles of analyte detection and regeneration. Such continuous and reliable SERS-based flow analysis accesses diverse applications from environmental monitoring to medical diagnostics. |
| 127. |  | Bing Ni, Dustin Vivod, Jonathan Avaro, Haoyuan Qi, Dirk Zahn, Xun Wang, Helmut Cölfen Reversible chirality inversion of an AuAgx-cysteine coordination polymer by pH change In: Nature Communications, vol. 15, no. 2042, 2024, (Reactive chiral systems have attracted much attention in biology, optoelectronics, and photonics; however, a comprehensive understanding of these systems remains incomplete. Here the authors show the reversible chirality of AuAgx-cys coordination polymers induced by pH changes.). @article{nokey,
title = {Reversible chirality inversion of an AuAgx-cysteine coordination polymer by pH change},
author = {Bing Ni and Dustin Vivod and Jonathan Avaro and Haoyuan Qi and Dirk Zahn and Xun Wang and Helmut Cölfen },
url = {https://www.nature.com/articles/s41467-024-45935-3.pdf},
doi = {10.1038/s41467-024-45935-3},
year = {2024},
date = {2024-03-06},
urldate = {2024-03-06},
journal = {Nature Communications},
volume = {15},
number = {2042},
abstract = {Responsive chiral systems have attracted considerable attention, given their potential for diverse applications in biology, optoelectronics, photonics, and related fields. Here we show the reversible chirality inversion of an AuAgx-cysteine (AuAgx-cys) coordination polymer (CP) by pH changes. The polymer can be obtained by mixing HAuCl4 and AgNO3 with L-cysteine (or D-cysteine) in appropriate proportions in H2O (or other surfactant solutions). Circular dichroism (CD) spectrum is used to record the strong optical activity of the AuAg0.06-L-cys enantiomer (denoted as L0.06), which can be switched to that of the corresponding D0.06 enantiomer by alkalization (final dispersion pH > 13) and can be switched back after neutralization (final dispersion pH <8). Multiple structural changes at different pH values (≈9.6, ≈13) are observed through UV-Vis and CD spectral measurements, as well as other controlled experiments. Exploration of the CP synthesis kinetics suggests that the covalent bond formation is rapid and then the conformation of the CP materials would continuously evolve. The reaction stoichiometry investigation shows that the formation of CP materials with chirality inversion behavior requires the balancing between different coordination and polymerization processes. This study provides insights into the potential of inorganic stereochemistry in developing promising functional materials.},
note = {Reactive chiral systems have attracted much attention in biology, optoelectronics, and photonics; however, a comprehensive understanding of these systems remains incomplete. Here the authors show the reversible chirality of AuAgx-cys coordination polymers induced by pH changes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Responsive chiral systems have attracted considerable attention, given their potential for diverse applications in biology, optoelectronics, photonics, and related fields. Here we show the reversible chirality inversion of an AuAgx-cysteine (AuAgx-cys) coordination polymer (CP) by pH changes. The polymer can be obtained by mixing HAuCl4 and AgNO3 with L-cysteine (or D-cysteine) in appropriate proportions in H2O (or other surfactant solutions). Circular dichroism (CD) spectrum is used to record the strong optical activity of the AuAg0.06-L-cys enantiomer (denoted as L0.06), which can be switched to that of the corresponding D0.06 enantiomer by alkalization (final dispersion pH > 13) and can be switched back after neutralization (final dispersion pH <8). Multiple structural changes at different pH values (≈9.6, ≈13) are observed through UV-Vis and CD spectral measurements, as well as other controlled experiments. Exploration of the CP synthesis kinetics suggests that the covalent bond formation is rapid and then the conformation of the CP materials would continuously evolve. The reaction stoichiometry investigation shows that the formation of CP materials with chirality inversion behavior requires the balancing between different coordination and polymerization processes. This study provides insights into the potential of inorganic stereochemistry in developing promising functional materials. |
| 126. | 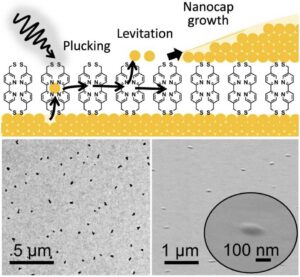 | Chenyang Guo, Philip Benzie, Shu Hu, Bart de Nijs, Ermanno Miele, Eoin Elliott, Rakesh Arul, Helen Benjamin, Grzegorz Dziechciarczyk, Reshma R. Rao, Mary P. Ryan, Jeremy J. Baumberg Extensive photochemical restructuring of molecule-metal surfaces under room light In: Nature Communications, vol. 15, no. 1928, 2024, (The nature of the molecule-metal interface is crucial for many technological applications. Here, the authors show that the photostability of the material can be sensitive to room light when coated with a single molecular layer, with implications for devices and processes.). @article{Guo2024,
title = {Extensive photochemical restructuring of molecule-metal surfaces under room light},
author = {Chenyang Guo and Philip Benzie and Shu Hu and Bart de Nijs and Ermanno Miele and Eoin Elliott and Rakesh Arul and Helen Benjamin and Grzegorz Dziechciarczyk and Reshma R. Rao and Mary P. Ryan and Jeremy J. Baumberg },
url = {https://www.nature.com/articles/s41467-024-46125-x.pdf},
doi = {10.1038/s41467-024-46125-x},
year = {2024},
date = {2024-03-02},
urldate = {2024-03-02},
journal = {Nature Communications},
volume = {15},
number = {1928},
abstract = {The molecule-metal interface is of paramount importance for many devices and processes, and directly involved in photocatalysis, molecular electronics, nanophotonics, and molecular (bio-)sensing. Here the photostability of this interface is shown to be sensitive even to room light levels for specific molecules and metals. Optical spectroscopy is used to track photoinduced migration of gold atoms when functionalised with different thiolated molecules that form uniform monolayers on Au. Nucleation and growth of characteristic surface metal nanostructures is observed from the light-driven adatoms. By watching the spectral shifts of optical modes from nanoparticles used to precoat these surfaces, we identify processes involved in the photo-migration mechanism and the chemical groups that facilitate it. This photosensitivity of the molecule-metal interface highlights the significance of optically induced surface reconstruction. In some catalytic contexts this can enhance activity, especially utilising atomically dispersed gold. Conversely, in electronic device applications such reconstructions introduce problematic aging effects.},
note = {The nature of the molecule-metal interface is crucial for many technological applications. Here, the authors show that the photostability of the material can be sensitive to room light when coated with a single molecular layer, with implications for devices and processes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The molecule-metal interface is of paramount importance for many devices and processes, and directly involved in photocatalysis, molecular electronics, nanophotonics, and molecular (bio-)sensing. Here the photostability of this interface is shown to be sensitive even to room light levels for specific molecules and metals. Optical spectroscopy is used to track photoinduced migration of gold atoms when functionalised with different thiolated molecules that form uniform monolayers on Au. Nucleation and growth of characteristic surface metal nanostructures is observed from the light-driven adatoms. By watching the spectral shifts of optical modes from nanoparticles used to precoat these surfaces, we identify processes involved in the photo-migration mechanism and the chemical groups that facilitate it. This photosensitivity of the molecule-metal interface highlights the significance of optically induced surface reconstruction. In some catalytic contexts this can enhance activity, especially utilising atomically dispersed gold. Conversely, in electronic device applications such reconstructions introduce problematic aging effects. |
| 125. | 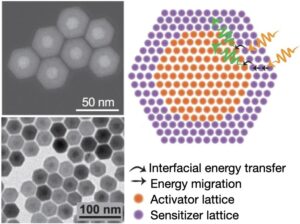 | Long Yan, Jinshu Huang, Zhengce An, Qinyuan Zhang, Bo Zhou Spatiotemporal control of photochromic upconversion through interfacial energy transfer In: Nature Communications, vol. 15, no. 1923, 2024, (Achieving spatiotemporal control of photochromic upconversion from a single lanthanide emitter remains challenging. Here, the authors present a conceptual model enabling such control of Er3+ photochromic upconversion via interfacial energy transfer in a core-shell nanostructure.). @article{Yan2024,
title = {Spatiotemporal control of photochromic upconversion through interfacial energy transfer},
author = {Long Yan and Jinshu Huang and Zhengce An and Qinyuan Zhang and Bo Zhou },
url = {https://www.nature.com/articles/s41467-024-46228-5.pdf},
doi = {10.1038/s41467-024-46228-5},
year = {2024},
date = {2024-03-01},
urldate = {2024-03-01},
journal = {Nature Communications},
volume = {15},
number = {1923},
abstract = {Dynamic control of multi-photon upconversion with rich and tunable emission colors is stimulating extensive interest in both fundamental research and frontier applications of lanthanide based materials. However, manipulating photochromic upconversion towards color-switchable emissions of a single lanthanide emitter is still challenging. Here, we report a conceptual model to realize the spatiotemporal control of upconversion dynamics and photochromic evolution of Er3+ through interfacial energy transfer (IET) in a core-shell nanostructure. The design of Yb sublattice sensitization interlayer, instead of regular Yb3+ doping, is able to raise the absorption capability of excitation energy and enhance the upconversion. We find that a nanoscale spatial manipulation of interfacial interactions between Er and Yb sublattices can further contribute to upconversion. Moreover, the red/green color-switchable upconversion of Er3+ is achieved through using the temporal modulation ways of non-steady-state excitation and time-gating technique. Our results allow for versatile designs and dynamic management of emission colors from luminescent materials and provide more chances for their frontier photonic applications such as optical anti-counterfeiting and speed monitoring.},
note = {Achieving spatiotemporal control of photochromic upconversion from a single lanthanide emitter remains challenging. Here, the authors present a conceptual model enabling such control of Er3+ photochromic upconversion via interfacial energy transfer in a core-shell nanostructure.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Dynamic control of multi-photon upconversion with rich and tunable emission colors is stimulating extensive interest in both fundamental research and frontier applications of lanthanide based materials. However, manipulating photochromic upconversion towards color-switchable emissions of a single lanthanide emitter is still challenging. Here, we report a conceptual model to realize the spatiotemporal control of upconversion dynamics and photochromic evolution of Er3+ through interfacial energy transfer (IET) in a core-shell nanostructure. The design of Yb sublattice sensitization interlayer, instead of regular Yb3+ doping, is able to raise the absorption capability of excitation energy and enhance the upconversion. We find that a nanoscale spatial manipulation of interfacial interactions between Er and Yb sublattices can further contribute to upconversion. Moreover, the red/green color-switchable upconversion of Er3+ is achieved through using the temporal modulation ways of non-steady-state excitation and time-gating technique. Our results allow for versatile designs and dynamic management of emission colors from luminescent materials and provide more chances for their frontier photonic applications such as optical anti-counterfeiting and speed monitoring. |
| 124. |  | Hayato Otsuka, Koki Urita, Nobutaka Honma, Takashi Kimuro, Yasushi Amako, Radovan Kukobat, Teresa J. Bandosz, Junzo Ukai, Isamu Moriguchi, Katsumi Kaneko Transient chemical and structural changes in graphene oxide during ripening In: Nature Communications, vol. 15, no. 1708, 2024, (Graphene oxide is in demand for various applications - however, this is complicated by changing physicochemical properties over time. Here, the authors show the intrinsic, metastable, and transient states of graphene oxide colloids upon ripening.
). @article{Otsuka2024,
title = {Transient chemical and structural changes in graphene oxide during ripening},
author = {Hayato Otsuka and Koki Urita and Nobutaka Honma and Takashi Kimuro and Yasushi Amako and Radovan Kukobat and Teresa J. Bandosz and Junzo Ukai and Isamu Moriguchi and Katsumi Kaneko },
url = {https://www.nature.com/articles/s41467-024-46083-4.pdf},
doi = {10.1038/s41467-024-46083-4},
year = {2024},
date = {2024-02-24},
urldate = {2024-02-24},
journal = {Nature Communications},
volume = {15},
number = {1708},
abstract = {Graphene oxide (GO)—the oxidized form of graphene—is actively studied in various fields, such as energy, electronic devices, separation of water, materials engineering, and medical technologies, owing to its fascinating physicochemical properties. One major drawback of GO is its instability, which leads to the difficulties in product management. A physicochemical understanding of the ever-changing nature of GO can remove the barrier for its growing applications. Here, we evidencde the presence of intrinsic, metastable and transient GO states upon ripening. The three GO states are identified using a pi-pi* transition peak of ultraviolet–visible absorption spectra and exhibit inherent magnetic and electrical properties. The presence of three states of GO is supported by the compositional changes of oxygen functional groups detected via X-ray photoelectron spectroscopy and structural information from X-ray diffraction analysis and transmission electron microscopy. Although intrinsic GO having a pi-pi* transition at 230.5 ± 0.5 nm is stable only for 5 days at 298 K, the intrinsic state can be stabilized by either storing GO dispersions below 255 K or by adding ammonium peroxydisulfate.},
note = {Graphene oxide is in demand for various applications - however, this is complicated by changing physicochemical properties over time. Here, the authors show the intrinsic, metastable, and transient states of graphene oxide colloids upon ripening.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Graphene oxide (GO)—the oxidized form of graphene—is actively studied in various fields, such as energy, electronic devices, separation of water, materials engineering, and medical technologies, owing to its fascinating physicochemical properties. One major drawback of GO is its instability, which leads to the difficulties in product management. A physicochemical understanding of the ever-changing nature of GO can remove the barrier for its growing applications. Here, we evidencde the presence of intrinsic, metastable and transient GO states upon ripening. The three GO states are identified using a pi-pi* transition peak of ultraviolet–visible absorption spectra and exhibit inherent magnetic and electrical properties. The presence of three states of GO is supported by the compositional changes of oxygen functional groups detected via X-ray photoelectron spectroscopy and structural information from X-ray diffraction analysis and transmission electron microscopy. Although intrinsic GO having a pi-pi* transition at 230.5 ± 0.5 nm is stable only for 5 days at 298 K, the intrinsic state can be stabilized by either storing GO dispersions below 255 K or by adding ammonium peroxydisulfate. |
| 123. | 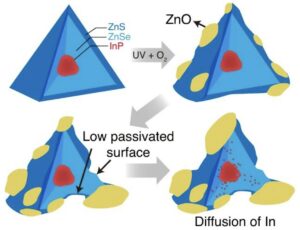 | Hayeon Baek, Sungsu Kang, Junyoung Heo, Soonmi Choi, Ran Kim, Kihyun Kim, Nari Ahn, Yeo-Geon Yoon, Taekjoon Lee, Jae Bok Chang, Kyung Sig Lee, Young-Gil Park, Jungwon Park Insights into structural defect formation in individual InP/ZnSe/ZnS quantum dots under UV oxidation In: Nature Communications, vol. 15, no. 1671, 2024, (InP/ZnSe/ZnS quantum dots (QDs) are promising candidates for advanced light-emitting diodes, but low emission efficiency due to oxidation hampers applications. Here, the authors provide insight into the structural defects that form on individual QDs during UV-facilitated oxidation. ). @article{Baek2024,
title = {Insights into structural defect formation in individual InP/ZnSe/ZnS quantum dots under UV oxidation},
author = {Hayeon Baek and Sungsu Kang and Junyoung Heo and Soonmi Choi and Ran Kim and Kihyun Kim and Nari Ahn and Yeo-Geon Yoon and Taekjoon Lee and Jae Bok Chang and Kyung Sig Lee and Young-Gil Park and Jungwon Park },
url = {https://www.nature.com/articles/s41467-024-45944-2.pdf},
doi = {10.1038/s41467-024-45944-2},
year = {2024},
date = {2024-02-23},
urldate = {2024-02-23},
journal = {Nature Communications},
volume = {15},
number = {1671},
abstract = {InP/ZnSe/ZnS quantum dots (QDs) stand as promising candidates for advancing QD-organic light-emitting diodes (QLED), but low emission efficiency due to their susceptibility to oxidation impedes applications. Structural defects play important roles in the emission efficiency degradation of QDs, but the formation mechanism of defects in oxidized QDs has been less investigated. Here, we investigated the impact of diverse structural defects formation on individual QDs and propagation during UV-facilitated oxidation using high-resolution (scanning) transmission electron microscopy. UV-facilitated oxidation of the QDs alters shell morphology by the formation of surface oxides, leaving ZnSe surfaces poorly passivated. Further oxidation leads to the formation of structural defects, such as dislocations, and induces strain at the oxide-QD interfaces, facilitating In diffusion from the QD core. These changes in the QD structures result in emission quenching. This study provides insight into the formation of structural defects through photo-oxidation, and their effects on emission properties of QDs.},
note = {InP/ZnSe/ZnS quantum dots (QDs) are promising candidates for advanced light-emitting diodes, but low emission efficiency due to oxidation hampers applications. Here, the authors provide insight into the structural defects that form on individual QDs during UV-facilitated oxidation. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
InP/ZnSe/ZnS quantum dots (QDs) stand as promising candidates for advancing QD-organic light-emitting diodes (QLED), but low emission efficiency due to their susceptibility to oxidation impedes applications. Structural defects play important roles in the emission efficiency degradation of QDs, but the formation mechanism of defects in oxidized QDs has been less investigated. Here, we investigated the impact of diverse structural defects formation on individual QDs and propagation during UV-facilitated oxidation using high-resolution (scanning) transmission electron microscopy. UV-facilitated oxidation of the QDs alters shell morphology by the formation of surface oxides, leaving ZnSe surfaces poorly passivated. Further oxidation leads to the formation of structural defects, such as dislocations, and induces strain at the oxide-QD interfaces, facilitating In diffusion from the QD core. These changes in the QD structures result in emission quenching. This study provides insight into the formation of structural defects through photo-oxidation, and their effects on emission properties of QDs. |
| 122. | 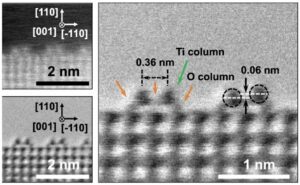 | Wentao Yuan, Bingwei Chen, Zhong-Kang Han, Ruiyang You, Ying Jiang, Rui Qi, Guanxing Li, Hanglong Wu, Maria Veronica Ganduglia-Pirovano, Yong Wang Direct in-situ insights into the asymmetric surface reconstruction of rutile TiO2 (110) In: Nature Communications, vol. 15, no. 1616, 2024, (The reconstruction of rutile TiO2 (110) impacts its surface chemistry and catalytic properties. Here, the authors offer a detailed understanding of the asymmetric surface reconstruction of TiO2 (110)-(1×2) through a combination of STEM and DFT calculations.). @article{Yuan2024,
title = {Direct in-situ insights into the asymmetric surface reconstruction of rutile TiO2 (110)},
author = {Wentao Yuan and Bingwei Chen and Zhong-Kang Han and Ruiyang You and Ying Jiang and Rui Qi and Guanxing Li and Hanglong Wu and Maria Veronica Ganduglia-Pirovano and Yong Wang },
url = {https://www.nature.com/articles/s41467-024-46011-6.pdf},
doi = {10.1038/s41467-024-46011-6},
year = {2024},
date = {2024-02-22},
urldate = {2024-02-22},
journal = {Nature Communications},
volume = {15},
number = {1616},
abstract = {The reconstruction of rutile TiO2 (110) holds significant importance as it profoundly influences the surface chemistry and catalytic properties of this widely used material in various applications, from photocatalysis to solar energy conversion. Here, we directly observe the asymmetric surface reconstruction of rutile TiO2 (110)-(1×2) with atomic-resolution using in situ spherical aberration-corrected scanning transmission electron microscopy. Density functional theory calculations were employed to complement the experimental observations. Our findings highlight the pivotal role played by repulsive electrostatic interaction among the small polarons −formed by excess electrons following the removal of neutral oxygen atoms− and the subsequent surface relaxations induced by these polarons. The emergence and disappearance of these asymmetric structures can be controlled by adjusting the oxygen partial pressure. This research provides a deeper understanding, prediction, and manipulation of the surface reconstructions of rutile TiO2 (110), holding implications for a diverse range of applications and technological advancements involving rutile-based materials.
},
note = {The reconstruction of rutile TiO2 (110) impacts its surface chemistry and catalytic properties. Here, the authors offer a detailed understanding of the asymmetric surface reconstruction of TiO2 (110)-(1×2) through a combination of STEM and DFT calculations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The reconstruction of rutile TiO2 (110) holds significant importance as it profoundly influences the surface chemistry and catalytic properties of this widely used material in various applications, from photocatalysis to solar energy conversion. Here, we directly observe the asymmetric surface reconstruction of rutile TiO2 (110)-(1×2) with atomic-resolution using in situ spherical aberration-corrected scanning transmission electron microscopy. Density functional theory calculations were employed to complement the experimental observations. Our findings highlight the pivotal role played by repulsive electrostatic interaction among the small polarons −formed by excess electrons following the removal of neutral oxygen atoms− and the subsequent surface relaxations induced by these polarons. The emergence and disappearance of these asymmetric structures can be controlled by adjusting the oxygen partial pressure. This research provides a deeper understanding, prediction, and manipulation of the surface reconstructions of rutile TiO2 (110), holding implications for a diverse range of applications and technological advancements involving rutile-based materials.
|
| 121. | 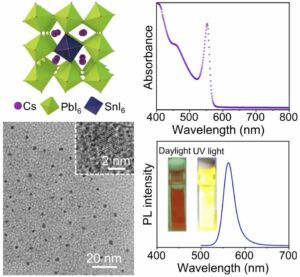 | Louwen Zhang, Hai Zhou, Yibo Chen, Zhimiao Zheng, Lishuai Huang, Chen Wang, Kailian Dong, Zhongqiang Hu, Weijun Ke, Guojia Fang Spontaneous crystallization of strongly confined CsSnxPb1-xI3 perovskite colloidal quantum dots at room temperature In: Nature Communications, vol. 15, no. 1609, 2024, (Photoactive pure-iodine all-inorganic colloidal perovskite quantum dots (QDs) are attractive for optoelectronic applications, however their synthesis at room temperature is challenging. Here the authors report a room temperature strongly confined strategy to synthesize CsSnxPb1-xI3 QDs.). @article{Zhang2024,
title = {Spontaneous crystallization of strongly confined CsSnxPb1-xI3 perovskite colloidal quantum dots at room temperature},
author = {Louwen Zhang and Hai Zhou and Yibo Chen and Zhimiao Zheng and Lishuai Huang and Chen Wang and Kailian Dong and Zhongqiang Hu and Weijun Ke and Guojia Fang },
url = {https://www.nature.com/articles/s41467-024-45945-1.pdf},
doi = {10.1038/s41467-024-45945-1},
year = {2024},
date = {2024-02-21},
urldate = {2024-02-21},
journal = {Nature Communications},
volume = {15},
number = {1609},
abstract = {The scalable and low-cost room temperature (RT) synthesis for pure-iodine all-inorganic perovskite colloidal quantum dots (QDs) is a challenge due to the phase transition induced by thermal unequilibrium. Here, we introduce a direct RT strongly confined spontaneous crystallization strategy in a Cs-deficient reaction system without polar solvents for synthesizing stable pure-iodine all-inorganic tin-lead (Sn-Pb) alloyed perovskite colloidal QDs, which exhibit bright yellow luminescence. By tuning the ratio of Cs/Pb precursors, the size confinement effect and optical band gap of the resultant CsSnxPb1-xI3 perovskite QDs can be well controlled. This strongly confined RT approach is universal for wider bandgap bromine- and chlorine-based all-inorganic and iodine-based hybrid perovskite QDs. The alloyed CsSn0.09Pb0.91I3 QDs show superior yellow emission properties with prolonged carrier lifetime and significantly increased colloidal stability compared to the pristine CsPbI3 QDs, which is enabled by strong size confinement, Sn2+ passivation and enhanced formation energy. These findings provide a RT size-stabilized synthesis pathway to achieve high-performance pure-iodine all-inorganic Sn-Pb mixed perovskite colloidal QDs for optoelectronic applications.},
note = {Photoactive pure-iodine all-inorganic colloidal perovskite quantum dots (QDs) are attractive for optoelectronic applications, however their synthesis at room temperature is challenging. Here the authors report a room temperature strongly confined strategy to synthesize CsSnxPb1-xI3 QDs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The scalable and low-cost room temperature (RT) synthesis for pure-iodine all-inorganic perovskite colloidal quantum dots (QDs) is a challenge due to the phase transition induced by thermal unequilibrium. Here, we introduce a direct RT strongly confined spontaneous crystallization strategy in a Cs-deficient reaction system without polar solvents for synthesizing stable pure-iodine all-inorganic tin-lead (Sn-Pb) alloyed perovskite colloidal QDs, which exhibit bright yellow luminescence. By tuning the ratio of Cs/Pb precursors, the size confinement effect and optical band gap of the resultant CsSnxPb1-xI3 perovskite QDs can be well controlled. This strongly confined RT approach is universal for wider bandgap bromine- and chlorine-based all-inorganic and iodine-based hybrid perovskite QDs. The alloyed CsSn0.09Pb0.91I3 QDs show superior yellow emission properties with prolonged carrier lifetime and significantly increased colloidal stability compared to the pristine CsPbI3 QDs, which is enabled by strong size confinement, Sn2+ passivation and enhanced formation energy. These findings provide a RT size-stabilized synthesis pathway to achieve high-performance pure-iodine all-inorganic Sn-Pb mixed perovskite colloidal QDs for optoelectronic applications. |
| 120. |  | Lei Lei, Minghao Yi, Yubin Wang, Youjie Hua, Junjie Zhang, Paras N. Prasad, Shiqing Xu Dual heterogeneous interfaces enhance X-ray excited persistent luminescence for low-dose 3D imaging In: Nature Communications, vol. 15, no. 1140, 2024, (High-resolution X-ray imaging requires a high radiation dose. Here, the authors achieve low-dose 3D imaging by increasing the XEPL intensity using a double-shell nanostructure with two heterogeneous interfaces.). @article{Lei2024,
title = {Dual heterogeneous interfaces enhance X-ray excited persistent luminescence for low-dose 3D imaging},
author = {Lei Lei and Minghao Yi and Yubin Wang and Youjie Hua and Junjie Zhang and Paras N. Prasad and Shiqing Xu },
url = {https://www.nature.com/articles/s41467-024-45390-0.pdf},
doi = {10.1038/s41467-024-45390-0},
year = {2024},
date = {2024-02-07},
urldate = {2024-02-07},
journal = {Nature Communications},
volume = {15},
number = {1140},
abstract = {Lanthanide-doped fluoride nanoparticles (NPs) showcase adjustable X-ray-excited persistent luminescence (XEPL), holding significant promise for applications in three-dimensional (3D) imaging through the creation of flexible X-ray detectors. However, a dangerous high X-ray irradiation dose rate and complicated heating procedure are required to generate efficient XEPL for high-resolution 3D imaging, which is attributed to a lack of strategies to significantly enhance the XEPL intensity. Here we report that the XEPL intensity of a series of lanthanide activators (Dy, Pr, Er, Tm, Gd, Tb) is greatly improved by constructing dual heterogeneous interfaces in a double-shell nanostructure. Mechanistic studies indicate that the employed core@shell@shell structure could not only passivate the surface quenchers to lower the non-radiative relaxation possibility, but also reduce the interfacial Frenkel defect formation energy leading to increase the trap concentration. By employing a NPs containing flexible film as the scintillation screen, the inside 3D electrical structure of a watch was clearly achieved based on the delayed XEPL imaging and 3D reconstruction procedure. We foresee that these findings will promote the development of advanced X-ray activated persistent fluoride NPs and offer opportunities for safer and more efficient X-ray imaging techniques in a number of scientific and practical areas.},
note = {High-resolution X-ray imaging requires a high radiation dose. Here, the authors achieve low-dose 3D imaging by increasing the XEPL intensity using a double-shell nanostructure with two heterogeneous interfaces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Lanthanide-doped fluoride nanoparticles (NPs) showcase adjustable X-ray-excited persistent luminescence (XEPL), holding significant promise for applications in three-dimensional (3D) imaging through the creation of flexible X-ray detectors. However, a dangerous high X-ray irradiation dose rate and complicated heating procedure are required to generate efficient XEPL for high-resolution 3D imaging, which is attributed to a lack of strategies to significantly enhance the XEPL intensity. Here we report that the XEPL intensity of a series of lanthanide activators (Dy, Pr, Er, Tm, Gd, Tb) is greatly improved by constructing dual heterogeneous interfaces in a double-shell nanostructure. Mechanistic studies indicate that the employed core@shell@shell structure could not only passivate the surface quenchers to lower the non-radiative relaxation possibility, but also reduce the interfacial Frenkel defect formation energy leading to increase the trap concentration. By employing a NPs containing flexible film as the scintillation screen, the inside 3D electrical structure of a watch was clearly achieved based on the delayed XEPL imaging and 3D reconstruction procedure. We foresee that these findings will promote the development of advanced X-ray activated persistent fluoride NPs and offer opportunities for safer and more efficient X-ray imaging techniques in a number of scientific and practical areas. |
| 119. |  | Yongsheng Sun, Yuzhen Wang, Weibin Chen, Qingquan Jiang, Dongdan Chen, Guoping Dong, Zhiguo Xia Rapid synthesis of phosphor-glass composites in seconds based on particle self-stabilization In: Nature Communications, vol. 15, no. 1033, 2024, (Phosphor-glass composites can serve as efficient and stable photonic converters, but their synthesis generally requires harsh and time-consuming procedures. Here, the authors report an alternative synthesis route that requires only a few seconds and is based on particle self-stabilization.). @article{Sun2024,
title = {Rapid synthesis of phosphor-glass composites in seconds based on particle self-stabilization},
author = {Yongsheng Sun and Yuzhen Wang and Weibin Chen and Qingquan Jiang and Dongdan Chen and Guoping Dong and Zhiguo Xia },
url = {https://www.nature.com/articles/s41467-024-45293-0.pdf},
doi = {10.1038/s41467-024-45293-0},
year = {2024},
date = {2024-02-05},
urldate = {2024-02-05},
journal = {Nature Communications},
volume = {15},
number = {1033},
abstract = {Phosphor-glass composites (PGC) are excellent candidates for highly efficient and stable photonic converters; however, their synthesis generally requires harsh procedures and long time, resulting in additional performance loss and energy consumption. Here we develop a rapid synthetic route to PGC within about 10 seconds, which enables uniform dispersion of Y3Al5O12:Ce3+ (YAG:Ce) phosphor particles through a particle self-stabilization model in molten tellurite glass. Thanks for good wettability between YAG:Ce micro-particles and tellurite glass melt, it creates an energy barrier of 6.94 × 105 zJ to prevent atomic-scale contact and sintering of particles in the melt. This in turn allows the generation of YAG:Ce-based PGC as attractive emitters with high quantum efficiency (98.4%) and absorption coefficient (86.8%) that can produce bright white light with luminous flux of 1227 lm and luminous efficiency of 276 lm W−1 under blue laser driving. This work shows a generalizable synthetic strategy for the development of functional glass composites.},
note = {Phosphor-glass composites can serve as efficient and stable photonic converters, but their synthesis generally requires harsh and time-consuming procedures. Here, the authors report an alternative synthesis route that requires only a few seconds and is based on particle self-stabilization.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Phosphor-glass composites (PGC) are excellent candidates for highly efficient and stable photonic converters; however, their synthesis generally requires harsh procedures and long time, resulting in additional performance loss and energy consumption. Here we develop a rapid synthetic route to PGC within about 10 seconds, which enables uniform dispersion of Y3Al5O12:Ce3+ (YAG:Ce) phosphor particles through a particle self-stabilization model in molten tellurite glass. Thanks for good wettability between YAG:Ce micro-particles and tellurite glass melt, it creates an energy barrier of 6.94 × 105 zJ to prevent atomic-scale contact and sintering of particles in the melt. This in turn allows the generation of YAG:Ce-based PGC as attractive emitters with high quantum efficiency (98.4%) and absorption coefficient (86.8%) that can produce bright white light with luminous flux of 1227 lm and luminous efficiency of 276 lm W−1 under blue laser driving. This work shows a generalizable synthetic strategy for the development of functional glass composites. |
| 118. |  | Ignacio Piquero-Zulaica, Eduardo Corral-Rascón, Xabier Diaz de Cerio, Alexander Riss, Biao Yang, Aran Garcia-Lekue, Mohammad A. Kher-Elden, Zakaria M. Abd El-Fattah, Shunpei Nobusue, Takahiro Kojima, Knud Seufert, Hiroshi Sakaguchi, Willi Auwärter, Johannes V. Barth Deceptive orbital confinement at edges and pores of carbon-based 1D and 2D nanoarchitectures In: Nature Communications, vol. 15, no. 1062, 2024, (The apparent electronic confinement at nanographene boundaries in scanning tunneling microscopy/spectroscopy is often misinterpreted. Here, the authors explain this phenomenon in terms of the decay of frontier orbitals and confinement at the edges of graphene nanoribbons and pores in nanoporous graphene.). @article{Piquero-Zulaica2024,
title = {Deceptive orbital confinement at edges and pores of carbon-based 1D and 2D nanoarchitectures},
author = {Ignacio Piquero-Zulaica and Eduardo Corral-Rascón and Xabier Diaz de Cerio and Alexander Riss and Biao Yang and Aran Garcia-Lekue and Mohammad A. Kher-Elden and Zakaria M. Abd El-Fattah and Shunpei Nobusue and Takahiro Kojima and Knud Seufert and Hiroshi Sakaguchi and Willi Auwärter and Johannes V. Barth},
url = {https://www.nature.com/articles/s41467-024-45138-w.pdf},
doi = {10.1038/s41467-024-45138-w},
year = {2024},
date = {2024-02-05},
urldate = {2024-02-05},
journal = {Nature Communications},
volume = {15},
number = {1062},
abstract = {The electronic structure defines the properties of graphene-based nanomaterials. Scanning tunneling microscopy/spectroscopy (STM/STS) experiments on graphene nanoribbons (GNRs), nanographenes, and nanoporous graphene (NPG) often determine an apparent electronic orbital confinement into the edges and nanopores, leading to dubious interpretations such as image potential states or super-atom molecular orbitals. We show that these measurements are subject to a wave function decay into the vacuum that masks the undisturbed electronic orbital shape. We use Au(111)-supported semiconducting gulf-type GNRs and NPGs as model systems fostering frontier orbitals that appear confined along the edges and nanopores in STS measurements. DFT calculations confirm that these states originate from valence and conduction bands. The deceptive electronic orbital confinement observed is caused by a loss of Fourier components, corresponding to states of high momentum. This effect can be generalized to other 1D and 2D carbon-based nanoarchitectures and is important for their use in catalysis and sensing applications.},
note = {The apparent electronic confinement at nanographene boundaries in scanning tunneling microscopy/spectroscopy is often misinterpreted. Here, the authors explain this phenomenon in terms of the decay of frontier orbitals and confinement at the edges of graphene nanoribbons and pores in nanoporous graphene.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The electronic structure defines the properties of graphene-based nanomaterials. Scanning tunneling microscopy/spectroscopy (STM/STS) experiments on graphene nanoribbons (GNRs), nanographenes, and nanoporous graphene (NPG) often determine an apparent electronic orbital confinement into the edges and nanopores, leading to dubious interpretations such as image potential states or super-atom molecular orbitals. We show that these measurements are subject to a wave function decay into the vacuum that masks the undisturbed electronic orbital shape. We use Au(111)-supported semiconducting gulf-type GNRs and NPGs as model systems fostering frontier orbitals that appear confined along the edges and nanopores in STS measurements. DFT calculations confirm that these states originate from valence and conduction bands. The deceptive electronic orbital confinement observed is caused by a loss of Fourier components, corresponding to states of high momentum. This effect can be generalized to other 1D and 2D carbon-based nanoarchitectures and is important for their use in catalysis and sensing applications. |
| 117. |  | Simon Wieland, Anja F. R. M. Ramsperger, Wolfgang Gross, Moritz Lehmann, Thomas Witzmann, Anja Caspari, Martin Obst, Stephan Gekle, Günter K. Auernhammer, Andreas Fery, Christian Laforsch, Holger Kress Nominally identical microplastic models differ greatly in their particle-cell interactions In: Nature Communications, vol. 15, no. 922, 2024, (Microplastics research is often based on commercial model particles. Here, the authors show that nominally identical particles may differ significantly in their properties and thus in their interactions with cells.). @article{nokey,
title = {Nominally identical microplastic models differ greatly in their particle-cell interactions},
author = {Simon Wieland and Anja F. R. M. Ramsperger and Wolfgang Gross and Moritz Lehmann and Thomas Witzmann and Anja Caspari and Martin Obst and Stephan Gekle and Günter K. Auernhammer and Andreas Fery and Christian Laforsch and Holger Kress},
url = {https://www.nature.com/articles/s41467-024-45281-4.pdf},
doi = {10.1038/s41467-024-45281-4},
year = {2024},
date = {2024-01-31},
urldate = {2024-01-31},
journal = {Nature Communications},
volume = {15},
number = {922},
abstract = {Due to the abundance of microplastics in the environment, research about its possible adverse effects is increasing exponentially. Most studies investigating the effect of microplastics on cells still rely on commercially available polystyrene microspheres. However, the choice of these model microplastic particles can affect the outcome of the studies, as even nominally identical model microplastics may interact differently with cells due to different surface properties such as the surface charge. Here, we show that nominally identical polystyrene microspheres from eight different manufacturers significantly differ in their ζ-potential, which is the electrical potential of a particle in a medium at its slipping plane. The ζ-potential of the polystyrene particles is additionally altered after environmental exposure. We developed a microfluidic microscopy platform to demonstrate that the ζ-potential determines particle-cell adhesion strength. Furthermore, we find that due to this effect, the ζ-potential also strongly determines the internalization of the microplastic particles into cells. Therefore, the ζ-potential can act as a proxy of microplastic-cell interactions and may govern adverse effects reported in various organisms exposed to microplastics.},
note = {Microplastics research is often based on commercial model particles. Here, the authors show that nominally identical particles may differ significantly in their properties and thus in their interactions with cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Due to the abundance of microplastics in the environment, research about its possible adverse effects is increasing exponentially. Most studies investigating the effect of microplastics on cells still rely on commercially available polystyrene microspheres. However, the choice of these model microplastic particles can affect the outcome of the studies, as even nominally identical model microplastics may interact differently with cells due to different surface properties such as the surface charge. Here, we show that nominally identical polystyrene microspheres from eight different manufacturers significantly differ in their ζ-potential, which is the electrical potential of a particle in a medium at its slipping plane. The ζ-potential of the polystyrene particles is additionally altered after environmental exposure. We developed a microfluidic microscopy platform to demonstrate that the ζ-potential determines particle-cell adhesion strength. Furthermore, we find that due to this effect, the ζ-potential also strongly determines the internalization of the microplastic particles into cells. Therefore, the ζ-potential can act as a proxy of microplastic-cell interactions and may govern adverse effects reported in various organisms exposed to microplastics. |
| 116. |  | Chenjie Dai, Shuai Wan, Zhe Li, Yangyang Shi, Shuang Zhang, Zhongyang Li Switchable unidirectional emissions from hydrogel gratings with integrated carbon quantum dots In: Nature Communications, vol. 15, no. 845, 2024, (Directional emission of photoluminescence is an emerging technique for light-emitting fields and nanophotonics. Here, the authors demonstrate a hydrogel grating with integrated quantum dots for switchable unidirectional emission tuning.). @article{Dai2024,
title = {Switchable unidirectional emissions from hydrogel gratings with integrated carbon quantum dots},
author = {Chenjie Dai and Shuai Wan and Zhe Li and Yangyang Shi and Shuang Zhang and Zhongyang Li },
url = {https://www.nature.com/articles/s41467-024-45284-1.pdf},
doi = {10.1038/s41467-024-45284-1},
year = {2024},
date = {2024-01-29},
urldate = {2024-01-29},
journal = {Nature Communications},
volume = {15},
number = {845},
abstract = {Directional emission of photoluminescence despite its incoherence is an attractive technique for light-emitting fields and nanophotonics. Optical metasurfaces provide a promising route for wavefront engineering at the subwavelength scale, enabling the feasibility of unidirectional emission. However, current directional emission strategies are mostly based on static metasurfaces, and it remains a challenge to achieve unidirectional emissions tuning with high performance. Here, we demonstrate quantum dots-hydrogel integrated gratings for actively switchable unidirectional emission with simultaneously a narrow divergence angle less than 1.5° and a large diffraction angle greater than 45°. We further demonstrate that the grating efficiency alteration leads to a more than 7-fold tuning of emission intensity at diffraction order due to the variation of hydrogel morphology subject to change in ambient humidity. Our proposed switchable emission strategy can promote technologies of active light-emitting devices for radiation control and optical imaging.},
note = {Directional emission of photoluminescence is an emerging technique for light-emitting fields and nanophotonics. Here, the authors demonstrate a hydrogel grating with integrated quantum dots for switchable unidirectional emission tuning.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Directional emission of photoluminescence despite its incoherence is an attractive technique for light-emitting fields and nanophotonics. Optical metasurfaces provide a promising route for wavefront engineering at the subwavelength scale, enabling the feasibility of unidirectional emission. However, current directional emission strategies are mostly based on static metasurfaces, and it remains a challenge to achieve unidirectional emissions tuning with high performance. Here, we demonstrate quantum dots-hydrogel integrated gratings for actively switchable unidirectional emission with simultaneously a narrow divergence angle less than 1.5° and a large diffraction angle greater than 45°. We further demonstrate that the grating efficiency alteration leads to a more than 7-fold tuning of emission intensity at diffraction order due to the variation of hydrogel morphology subject to change in ambient humidity. Our proposed switchable emission strategy can promote technologies of active light-emitting devices for radiation control and optical imaging. |
| 115. |  | Vito Coviello, Denis Badocco, Paolo Pastore, Martina Fracchia, Paolo Ghigna, Alessandro Martucci, Daniel Forrer, Vincenzo Amendola Accurate prediction of the optical properties of nanoalloys with both plasmonic and magnetic elements In: Nature Communications, vol. 15, no. 834, 2024, (The optical properties of nanoalloys are complex and difficult to describe. Here, the authors use density functional and Mie theory to calculate the extinction of Au-Co and other nanoalloys of interest for quantum optics, magnetooptics, catalysis, and metamaterials.). @article{nokey,
title = {Accurate prediction of the optical properties of nanoalloys with both plasmonic and magnetic elements},
author = {Vito Coviello and Denis Badocco and Paolo Pastore and Martina Fracchia and Paolo Ghigna and Alessandro Martucci and Daniel Forrer and Vincenzo Amendola },
url = {https://www.nature.com/articles/s41467-024-45137-x.pdf},
doi = {10.1038/s41467-024-45137-x},
year = {2024},
date = {2024-01-27},
urldate = {2024-01-27},
journal = {Nature Communications},
volume = {15},
number = {834},
abstract = {The alloying process plays a pivotal role in the development of advanced multifunctional plasmonic materials within the realm of modern nanotechnology. However, accurate in silico predictions are only available for metal clusters of just a few nanometers, while the support of modelling is required to navigate the broad landscape of components, structures and stoichiometry of plasmonic nanoalloys regardless of their size. Here we report on the accurate calculation and conceptual understanding of the optical properties of metastable alloys of both plasmonic (Au) and magnetic (Co) elements obtained through a tailored laser synthesis procedure. The model is based on the density functional theory calculation of the dielectric function with the Hubbard-corrected local density approximation, the correction for intrinsic size effects and use of classical electrodynamics. This approach is built to manage critical aspects in modelling of real samples, as spin polarization effects due to magnetic elements, short-range order variability, and size heterogeneity. The method provides accurate results also for other magnetic-plasmonic (Au-Fe) and typical plasmonic (Au-Ag) nanoalloys, thus being available for the investigation of several other nanomaterials waiting for assessment and exploitation in fundamental sectors such as quantum optics, magneto-optics, magneto-plasmonics, metamaterials, chiral catalysis and plasmon-enhanced catalysis.},
note = {The optical properties of nanoalloys are complex and difficult to describe. Here, the authors use density functional and Mie theory to calculate the extinction of Au-Co and other nanoalloys of interest for quantum optics, magnetooptics, catalysis, and metamaterials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The alloying process plays a pivotal role in the development of advanced multifunctional plasmonic materials within the realm of modern nanotechnology. However, accurate in silico predictions are only available for metal clusters of just a few nanometers, while the support of modelling is required to navigate the broad landscape of components, structures and stoichiometry of plasmonic nanoalloys regardless of their size. Here we report on the accurate calculation and conceptual understanding of the optical properties of metastable alloys of both plasmonic (Au) and magnetic (Co) elements obtained through a tailored laser synthesis procedure. The model is based on the density functional theory calculation of the dielectric function with the Hubbard-corrected local density approximation, the correction for intrinsic size effects and use of classical electrodynamics. This approach is built to manage critical aspects in modelling of real samples, as spin polarization effects due to magnetic elements, short-range order variability, and size heterogeneity. The method provides accurate results also for other magnetic-plasmonic (Au-Fe) and typical plasmonic (Au-Ag) nanoalloys, thus being available for the investigation of several other nanomaterials waiting for assessment and exploitation in fundamental sectors such as quantum optics, magneto-optics, magneto-plasmonics, metamaterials, chiral catalysis and plasmon-enhanced catalysis. |
| 114. |  | Simon Settele, C. Alexander Schrage, Sebastian Jung, Elena Michel, Han Li, Benjamin S. Flavel, A. Stephen K. Hashmi, Sebastian Kruss, Jana Zaumseil Ratiometric fluorescent sensing of pyrophosphate with sp³-functionalized single-walled carbon nanotubes In: Nature Communications, vol. 14, no. 706, 2024, (Inorganic pyrophosphate is a key molecule in many biological processes. Here, the authors develop an optical sensor that enables its ratiometric detection in the near infrared with functionalized single-walled carbon nanotubes.). @article{nokey,
title = {Ratiometric fluorescent sensing of pyrophosphate with sp³-functionalized single-walled carbon nanotubes},
author = {Simon Settele and C. Alexander Schrage and Sebastian Jung and Elena Michel and Han Li and Benjamin S. Flavel and A. Stephen K. Hashmi and Sebastian Kruss and Jana Zaumseil },
url = {https://www.nature.com/articles/s41467-024-45052-1.pdf},
doi = {10.1038/s41467-024-45052-1},
year = {2024},
date = {2024-01-24},
urldate = {2024-01-24},
journal = {Nature Communications},
volume = {14},
number = {706},
abstract = {Inorganic pyrophosphate is a key molecule in many biological processes from DNA synthesis to cell metabolism. Here we introduce sp3-functionalized (6,5) single-walled carbon nanotubes (SWNTs) with red-shifted defect emission as near-infrared luminescent probes for the optical detection and quantification of inorganic pyrophosphate. The sensing scheme is based on the immobilization of Cu2+ ions on the SWNT surface promoted by coordination to covalently attached aryl alkyne groups and a triazole complex. The presence of Cu2+ ions on the SWNT surface causes fluorescence quenching via photoinduced electron transfer, which is reversed by copper-complexing analytes such as pyrophosphate. The differences in the fluorescence response of sp3-defect to pristine nanotube emission enables reproducible ratiometric measurements in a wide concentration window. Biocompatible, phospholipid-polyethylene glycol-coated SWNTs with such sp3 defects are employed for the detection of pyrophosphate in cell lysate and for monitoring the progress of DNA synthesis in a polymerase chain reaction. This robust ratiometric and near-infrared luminescent probe for pyrophosphate may serve as a starting point for the rational design of nanotube-based biosensors.},
note = {Inorganic pyrophosphate is a key molecule in many biological processes. Here, the authors develop an optical sensor that enables its ratiometric detection in the near infrared with functionalized single-walled carbon nanotubes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Inorganic pyrophosphate is a key molecule in many biological processes from DNA synthesis to cell metabolism. Here we introduce sp3-functionalized (6,5) single-walled carbon nanotubes (SWNTs) with red-shifted defect emission as near-infrared luminescent probes for the optical detection and quantification of inorganic pyrophosphate. The sensing scheme is based on the immobilization of Cu2+ ions on the SWNT surface promoted by coordination to covalently attached aryl alkyne groups and a triazole complex. The presence of Cu2+ ions on the SWNT surface causes fluorescence quenching via photoinduced electron transfer, which is reversed by copper-complexing analytes such as pyrophosphate. The differences in the fluorescence response of sp3-defect to pristine nanotube emission enables reproducible ratiometric measurements in a wide concentration window. Biocompatible, phospholipid-polyethylene glycol-coated SWNTs with such sp3 defects are employed for the detection of pyrophosphate in cell lysate and for monitoring the progress of DNA synthesis in a polymerase chain reaction. This robust ratiometric and near-infrared luminescent probe for pyrophosphate may serve as a starting point for the rational design of nanotube-based biosensors. |
| 113. | ![Synthesis of inter-[60]fullerene conjugates with inherent chirality](https://christiankuttner.de/wp-content/uploads/2024/02/TOC113-300x253.jpeg) | Yoshifumi Hashikawa, Shu Okamoto, Yasujiro Murata Synthesis of inter-[60]fullerene conjugates with inherent chirality In: Nature Communications, vol. 15, no. 514, 2024, (Inter-fullerene conjugates are non-naturally occurring carbon allotropes. Here the authors report on the chemical synthesis and solid-state structure of three inter-[60]fullerene hybrids with inherent chirality.
). @article{nokey,
title = {Synthesis of inter-[60]fullerene conjugates with inherent chirality},
author = {Yoshifumi Hashikawa and Shu Okamoto and Yasujiro Murata },
url = {https://www.nature.com/articles/s41467-024-44834-x.pdf},
doi = {10.1038/s41467-024-44834-x},
year = {2024},
date = {2024-01-15},
urldate = {2024-01-15},
journal = {Nature Communications},
volume = {15},
number = {514},
abstract = {Coalescence of [60]fullerenes potentially produces hypothetical nanocarbon assemblies with non-naturally occurring topologies. Since the discovery of [60]fullerene in 1985, coalesced [60]fullerene oligomers have only been observed as transient species by transmission electron microscopy during an oligomerization process under a high electron acceleration voltage. Herein, we showcase the rational synthesis of covalent assemblies consisting of inherently chiral open-[60]fullerenes. The crystallographic analyses unveiled double-caged structures of non-conjugated and conjugated inter-[60]fullerene hybrids, in which the two [60]fullerene cages are bounds to each other through a covalent linkage. The former one further assembles via a heterochiral recognition so that four carbon cages are arranged in a tetrahedral manner both in solution and solid state. Reflecting radially-conjugated double π-surface nature, the inter-[60]fullerene conjugate exhibits strong electronic communication in its reduced states, intense absorption behavior, and chiroptical activity with a dissymmetry factor of 0.21 (at 674 nm) which breaks the record for known chiral organic molecules.},
note = {Inter-fullerene conjugates are non-naturally occurring carbon allotropes. Here the authors report on the chemical synthesis and solid-state structure of three inter-[60]fullerene hybrids with inherent chirality.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coalescence of [60]fullerenes potentially produces hypothetical nanocarbon assemblies with non-naturally occurring topologies. Since the discovery of [60]fullerene in 1985, coalesced [60]fullerene oligomers have only been observed as transient species by transmission electron microscopy during an oligomerization process under a high electron acceleration voltage. Herein, we showcase the rational synthesis of covalent assemblies consisting of inherently chiral open-[60]fullerenes. The crystallographic analyses unveiled double-caged structures of non-conjugated and conjugated inter-[60]fullerene hybrids, in which the two [60]fullerene cages are bounds to each other through a covalent linkage. The former one further assembles via a heterochiral recognition so that four carbon cages are arranged in a tetrahedral manner both in solution and solid state. Reflecting radially-conjugated double π-surface nature, the inter-[60]fullerene conjugate exhibits strong electronic communication in its reduced states, intense absorption behavior, and chiroptical activity with a dissymmetry factor of 0.21 (at 674 nm) which breaks the record for known chiral organic molecules. |
| 112. |  | Cian Gabbett, Luke Doolan, Kevin Synnatschke, Laura Gambini, Emmet Coleman, Adam G. Kelly, Shixin Liu, Eoin Caffrey, Jose Munuera, Catriona Murphy, Stefano Sanvito, Lewys Jones, Jonathan N. Coleman
Quantitative analysis of printed nanostructured networks using high-resolution 3D FIB-SEM nanotomography In: Nature Communications, vol. 15, no. 278, 2024, (The physical properties of devices made of printed nanosheets and nanowires are determined by their intrinsic nanostructured network morphology. Here, the authors use FIB-SEM nanotomography to quantitatively analyze printed nanostructured networks via 3D reconstructions.). @article{nokey,
title = {Quantitative analysis of printed nanostructured networks using high-resolution 3D FIB-SEM nanotomography},
author = {Cian Gabbett and Luke Doolan and Kevin Synnatschke and Laura Gambini and Emmet Coleman and Adam G. Kelly and Shixin Liu and Eoin Caffrey and Jose Munuera and Catriona Murphy and Stefano Sanvito and Lewys Jones and Jonathan N. Coleman
},
url = {https://www.nature.com/articles/s41467-023-44450-1.pdf},
doi = {10.1038/s41467-023-44450-1},
year = {2024},
date = {2024-01-04},
urldate = {2024-01-04},
journal = {Nature Communications},
volume = {15},
number = {278},
abstract = {Networks of solution-processed nanomaterials are becoming increasingly important across applications in electronics, sensing and energy storage/generation. Although the physical properties of these devices are often completely dominated by network morphology, the network structure itself remains difficult to interrogate. Here, we utilise focused ion beam – scanning electron microscopy nanotomography (FIB-SEM-NT) to quantitatively characterise the morphology of printed nanostructured networks and their devices using nanometre-resolution 3D images. The influence of nanosheet/nanowire size on network structure in printed films of graphene, WS2 and silver nanosheets (AgNSs), as well as networks of silver nanowires (AgNWs), is investigated. We present a comprehensive toolkit to extract morphological characteristics including network porosity, tortuosity, specific surface area, pore dimensions and nanosheet orientation, which we link to network resistivity. By extending this technique to interrogate the structure and interfaces within printed vertical heterostacks, we demonstrate the potential of this technique for device characterisation and optimisation.},
note = {The physical properties of devices made of printed nanosheets and nanowires are determined by their intrinsic nanostructured network morphology. Here, the authors use FIB-SEM nanotomography to quantitatively analyze printed nanostructured networks via 3D reconstructions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Networks of solution-processed nanomaterials are becoming increasingly important across applications in electronics, sensing and energy storage/generation. Although the physical properties of these devices are often completely dominated by network morphology, the network structure itself remains difficult to interrogate. Here, we utilise focused ion beam – scanning electron microscopy nanotomography (FIB-SEM-NT) to quantitatively characterise the morphology of printed nanostructured networks and their devices using nanometre-resolution 3D images. The influence of nanosheet/nanowire size on network structure in printed films of graphene, WS2 and silver nanosheets (AgNSs), as well as networks of silver nanowires (AgNWs), is investigated. We present a comprehensive toolkit to extract morphological characteristics including network porosity, tortuosity, specific surface area, pore dimensions and nanosheet orientation, which we link to network resistivity. By extending this technique to interrogate the structure and interfaces within printed vertical heterostacks, we demonstrate the potential of this technique for device characterisation and optimisation. |
| 111. |  | Han Xue, Linhai Li, Yiqun Wang, Youhua Lu, Kai Cui, Zhiyuan He, Guoying Bai, Jie Liu, Xin Zhou, Jianjun Wang Probing the critical nucleus size in tetrahydrofuran clathrate hydrate formation using surface-anchored nanoparticles In: Nature Communications, vol. 15, no. 157, 2024, (The critical nucleus, which considered a key step in the formation of clathrate hydrates, has not yet been empirically confirmed. Here, the authors probe the critical nucleus size in clathrate formation of tetrahydrofuran and thus provide mechanistic insights.). @article{nokey,
title = {Probing the critical nucleus size in tetrahydrofuran clathrate hydrate formation using surface-anchored nanoparticles},
author = {Han Xue and Linhai Li and Yiqun Wang and Youhua Lu and Kai Cui and Zhiyuan He and Guoying Bai and Jie Liu and Xin Zhou and Jianjun Wang },
url = {https://www.nature.com/articles/s41467-023-44378-6.pdf},
doi = {10.1038/s41467-023-44378-6},
year = {2024},
date = {2024-01-02},
urldate = {2024-01-02},
journal = {Nature Communications},
volume = {15},
number = {157},
abstract = {Controlling the formation of clathrate hydrates is crucial for advancing hydrate-based technologies. However, the microscopic mechanism underlying clathrate hydrate formation through nucleation remains poorly elucidated. Specifically, the critical nucleus, theorized as a pivotal step in nucleation, lacks empirical validation. Here, we report uniform nanoparticles, e.g., graphene oxide (GO) nanosheets and gold or silver nanocubes with controlled sizes, as nanoprobes to experimentally measure the size of the critical nucleus of tetrahydrofuran (THF) clathrate hydrate formation. The capability of the nanoparticles in facilitating THF clathrate hydrate nucleation displays generally an abrupt change at a nanoparticle-size-determined specific supercooling. It is revealed that the free-energy barrier shows an abrupt change when the nanoparticles have an approximately the same size as that of the critical nucleus. Thus, it is inferred that THF clathrate hydrate nucleation involves the creation of a critical nucleus with its size being inversely proportional to the supercooling. By proving the existence and determining the supercooling-dependent size of the critical nucleus of the THF clathrate hydrates, this work brings insights in the microscopic pictures of the clathrate hydrate nucleation.},
note = {The critical nucleus, which considered a key step in the formation of clathrate hydrates, has not yet been empirically confirmed. Here, the authors probe the critical nucleus size in clathrate formation of tetrahydrofuran and thus provide mechanistic insights.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Controlling the formation of clathrate hydrates is crucial for advancing hydrate-based technologies. However, the microscopic mechanism underlying clathrate hydrate formation through nucleation remains poorly elucidated. Specifically, the critical nucleus, theorized as a pivotal step in nucleation, lacks empirical validation. Here, we report uniform nanoparticles, e.g., graphene oxide (GO) nanosheets and gold or silver nanocubes with controlled sizes, as nanoprobes to experimentally measure the size of the critical nucleus of tetrahydrofuran (THF) clathrate hydrate formation. The capability of the nanoparticles in facilitating THF clathrate hydrate nucleation displays generally an abrupt change at a nanoparticle-size-determined specific supercooling. It is revealed that the free-energy barrier shows an abrupt change when the nanoparticles have an approximately the same size as that of the critical nucleus. Thus, it is inferred that THF clathrate hydrate nucleation involves the creation of a critical nucleus with its size being inversely proportional to the supercooling. By proving the existence and determining the supercooling-dependent size of the critical nucleus of the THF clathrate hydrates, this work brings insights in the microscopic pictures of the clathrate hydrate nucleation. |
| 110. |  | Michael T. Robo, Ryan L. Hayes, Xinqiang Ding, Brian Pulawski, Jonah Z. Vilseck Fast free energy estimates from λ-dynamics with bias-updated Gibbs sampling In: Nature Communications, vol. 14, no. 8515, 2023, (Calculations of relative binding free energy are crucial for lead optimization in structure-based drug design, but classical methods are computationally expensive. Here, the authors describe a more efficient method for calculating the free energy that is as accurate as thermodynamic integration.). @article{nokey,
title = {Fast free energy estimates from λ-dynamics with bias-updated Gibbs sampling},
author = {Michael T. Robo and Ryan L. Hayes and Xinqiang Ding and Brian Pulawski and Jonah Z. Vilseck },
url = {https://www.nature.com/articles/s41467-023-44208-9.pdf},
doi = {10.1038/s41467-023-44208-9},
year = {2023},
date = {2023-12-21},
urldate = {2023-12-21},
journal = {Nature Communications},
volume = {14},
number = {8515},
abstract = {Relative binding free energy calculations have become an integral computational tool for lead optimization in structure-based drug design. Classical alchemical methods, including free energy perturbation or thermodynamic integration, compute relative free energy differences by transforming one molecule into another. However, these methods have high operational costs due to the need to perform many pairwise perturbations independently. To reduce costs and accelerate molecular design workflows, we present a method called λ-dynamics with bias-updated Gibbs sampling. This method uses dynamic biases to continuously sample between multiple ligand analogues collectively within a single simulation. We show that many relative binding free energies can be determined quickly with this approach without compromising accuracy. For five benchmark systems, agreement to experiment is high, with root mean square errors near or below 1.0 kcal mol−1. Free energy results are consistent with other computational approaches and within statistical noise of both methods (0.4 kcal mol−1 or less). Notably, large efficiency gains over thermodynamic integration of 18–66-fold for small perturbations and 100–200-fold for whole aromatic ring substitutions are observed. The rapid determination of relative binding free energies will enable larger chemical spaces to be more readily explored and structure-based drug design to be accelerated.},
note = {Calculations of relative binding free energy are crucial for lead optimization in structure-based drug design, but classical methods are computationally expensive. Here, the authors describe a more efficient method for calculating the free energy that is as accurate as thermodynamic integration.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Relative binding free energy calculations have become an integral computational tool for lead optimization in structure-based drug design. Classical alchemical methods, including free energy perturbation or thermodynamic integration, compute relative free energy differences by transforming one molecule into another. However, these methods have high operational costs due to the need to perform many pairwise perturbations independently. To reduce costs and accelerate molecular design workflows, we present a method called λ-dynamics with bias-updated Gibbs sampling. This method uses dynamic biases to continuously sample between multiple ligand analogues collectively within a single simulation. We show that many relative binding free energies can be determined quickly with this approach without compromising accuracy. For five benchmark systems, agreement to experiment is high, with root mean square errors near or below 1.0 kcal mol−1. Free energy results are consistent with other computational approaches and within statistical noise of both methods (0.4 kcal mol−1 or less). Notably, large efficiency gains over thermodynamic integration of 18–66-fold for small perturbations and 100–200-fold for whole aromatic ring substitutions are observed. The rapid determination of relative binding free energies will enable larger chemical spaces to be more readily explored and structure-based drug design to be accelerated. |
| 109. |  | Tianran Zhang, Dengping Lyu, Wei Xu, Xuan Feng, Ran Ni, Yufeng Wang Janus particles with tunable patch symmetry and their assembly into chiral colloidal clusters In: Nature Communications, vol. 14, no. 8494, 2023, (Janus particles commonly exhibit a high-symmetry patch, constraining the range of possible assemblies. Here, the authors devise a synthetic approach to fine-tune the patch symmetry in Janus particles and showcase the assembly of these particles into chiral colloidal clusters.). @article{nokey,
title = {Janus particles with tunable patch symmetry and their assembly into chiral colloidal clusters},
author = {Tianran Zhang and Dengping Lyu and Wei Xu and Xuan Feng and Ran Ni and Yufeng Wang },
url = {https://www.nature.com/articles/s41467-023-44154-6.pdf},
doi = {10.1038/s41467-023-44154-6},
year = {2023},
date = {2023-12-20},
urldate = {2023-12-20},
journal = {Nature Communications},
volume = {14},
number = {8494},
abstract = {Janus particles, which have an attractive patch on the otherwise repulsive surface, have been commonly employed for anisotropic colloidal assembly. While current methods of particle synthesis allow for control over the patch size, they are generally limited to producing dome-shaped patches with a high symmetry (C∞). Here, we report on the synthesis of Janus particles with patches of various tunable shapes, having reduced symmetries ranging from C2v to C3v and C4v. The Janus particles are synthesized by partial encapsulation of an octahedral metal-organic framework particle (UiO-66) in a polymer matrix. The extent of encapsulation is precisely regulated by a stepwise, asymmetric dewetting process that exposes selected facets of the UiO-66 particle. With depletion interaction, the Janus particles spontaneously assemble into colloidal clusters reflecting the particles’ shapes and patch symmetries. We observe the formation of chiral structures, whereby chirality emerges from achiral building blocks. With the ability to encode symmetry and directional bonding information, our strategy could give access to more complex colloidal superstructures through assembly.},
note = {Janus particles commonly exhibit a high-symmetry patch, constraining the range of possible assemblies. Here, the authors devise a synthetic approach to fine-tune the patch symmetry in Janus particles and showcase the assembly of these particles into chiral colloidal clusters.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Janus particles, which have an attractive patch on the otherwise repulsive surface, have been commonly employed for anisotropic colloidal assembly. While current methods of particle synthesis allow for control over the patch size, they are generally limited to producing dome-shaped patches with a high symmetry (C∞). Here, we report on the synthesis of Janus particles with patches of various tunable shapes, having reduced symmetries ranging from C2v to C3v and C4v. The Janus particles are synthesized by partial encapsulation of an octahedral metal-organic framework particle (UiO-66) in a polymer matrix. The extent of encapsulation is precisely regulated by a stepwise, asymmetric dewetting process that exposes selected facets of the UiO-66 particle. With depletion interaction, the Janus particles spontaneously assemble into colloidal clusters reflecting the particles’ shapes and patch symmetries. We observe the formation of chiral structures, whereby chirality emerges from achiral building blocks. With the ability to encode symmetry and directional bonding information, our strategy could give access to more complex colloidal superstructures through assembly. |
| 108. |  | Fenghua Zhang, Zhong Li, Xun Wang Mechanically tunable organogels from highly charged polyoxometalate clusters loaded with fluorescent dyes In: Nature Communications, vol. 14, no. 8327, 2023, (Functionalizing inorganic organogels is a challenge and can have a negative impact on their mechanical properties. Here, the authors present nanowire-based organogels based on highly charged polyoxometalate cluster units that are mechanically tuned and loaded with fluorescent dyes.). @article{nokey,
title = {Mechanically tunable organogels from highly charged polyoxometalate clusters loaded with fluorescent dyes},
author = {Fenghua Zhang and Zhong Li and Xun Wang },
url = {https://www.nature.com/articles/s41467-023-43989-3.pdf},
doi = {10.1038/s41467-023-43989-3},
year = {2023},
date = {2023-12-14},
urldate = {2023-12-14},
journal = {Nature Communications},
volume = {14},
number = {8327},
abstract = {Inorganic nanowires-based organogel, a class of emerging organogel with convenient preparation, recyclability, and excellent mechanical properties, is in its infancy. Solidifying and functionalizing nanowires-based organogels by designing the gelator structure remains challenging. Here, we fabricate Ca2-P2W16 and Ca2-P2W15M nanowires utilizing highly charged [Ca2P2W16O60]10− and [Ca2P2W15MO60]14−/13− cluster units, respectively, which are then employed for preparing organogels. The mechanical performance and stability of prepared organogels are improved due to the enhanced interactions between nanowires and locked organic molecules. Compressive stress and tensile stress of Ca2-P2W16 nanowires-based organogel reach 34.5 and 29.0 kPa, respectively. The critical gel concentration of Ca2-P2W16 nanowires is as low as 0.28%. Single-molecule force spectroscopy confirms that the connections between cluster units and linkers can regulate the flexibility of nanowires. Furthermore, the incorporation of fluorophores into the organogels adds fluorescence properties. This work reveals the relationships between the microstructures of inorganic gelators and the properties of organogels, guiding the synthesis of high-performance and functional organogels.},
note = {Functionalizing inorganic organogels is a challenge and can have a negative impact on their mechanical properties. Here, the authors present nanowire-based organogels based on highly charged polyoxometalate cluster units that are mechanically tuned and loaded with fluorescent dyes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Inorganic nanowires-based organogel, a class of emerging organogel with convenient preparation, recyclability, and excellent mechanical properties, is in its infancy. Solidifying and functionalizing nanowires-based organogels by designing the gelator structure remains challenging. Here, we fabricate Ca2-P2W16 and Ca2-P2W15M nanowires utilizing highly charged [Ca2P2W16O60]10− and [Ca2P2W15MO60]14−/13− cluster units, respectively, which are then employed for preparing organogels. The mechanical performance and stability of prepared organogels are improved due to the enhanced interactions between nanowires and locked organic molecules. Compressive stress and tensile stress of Ca2-P2W16 nanowires-based organogel reach 34.5 and 29.0 kPa, respectively. The critical gel concentration of Ca2-P2W16 nanowires is as low as 0.28%. Single-molecule force spectroscopy confirms that the connections between cluster units and linkers can regulate the flexibility of nanowires. Furthermore, the incorporation of fluorophores into the organogels adds fluorescence properties. This work reveals the relationships between the microstructures of inorganic gelators and the properties of organogels, guiding the synthesis of high-performance and functional organogels. |
| 107. |  | Vibhuti Rai, Nico Balzer, Gabriel Derenbach, Christof Holzer, Marcel Mayor, Wulf Wulfhekel, Lukas Gerhard, Michal Valášek Hot luminescence from single-molecule chromophores electrically and mechanically self-decoupled by tripodal scaffolds In: Nature Communications, vol. 14, no. 8253, 2023, (A fundamental challenge for molecular electronics is the change in photophysical properties of molecules upon direct electrical contact. Here, the authors observe hot luminescence emitted by single-molecule chromophores that are electrically and mechanically self-decoupled by a tripodal scaffold.). @article{nokey,
title = {Hot luminescence from single-molecule chromophores electrically and mechanically self-decoupled by tripodal scaffolds},
author = {Vibhuti Rai and Nico Balzer and Gabriel Derenbach and Christof Holzer and Marcel Mayor and Wulf Wulfhekel and Lukas Gerhard and Michal Valášek },
url = {https://www.nature.com/articles/s41467-023-43948-y.pdf},
doi = {10.1038/s41467-023-43948-y},
year = {2023},
date = {2023-12-12},
urldate = {2023-12-12},
journal = {Nature Communications},
volume = {14},
number = {8253},
abstract = {Control over the electrical contact to an individual molecule is one of the biggest challenges in molecular optoelectronics. The mounting of individual chromophores on extended tripodal scaffolds enables both efficient electrical and mechanical decoupling of individual chromophores from metallic leads. Core-substituted naphthalene diimides fixed perpendicular to a gold substrate by a covalently attached extended tripod display high stability with well-defined and efficient electroluminescence down to the single-molecule level. The molecularly controlled spatial arrangement balances the electric conduction for electroluminescence and the insulation to avoid non-radiative carrier recombination, enabling the spectrally and spatially resolved electroluminescence of individual self-decoupled chromophores in a scanning tunneling microscope. Hot luminescence bands are even visible in single self-decoupled chromophores, documenting the mechanical decoupling between the vibrons of the chromophore and the substrate.},
note = {A fundamental challenge for molecular electronics is the change in photophysical properties of molecules upon direct electrical contact. Here, the authors observe hot luminescence emitted by single-molecule chromophores that are electrically and mechanically self-decoupled by a tripodal scaffold.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Control over the electrical contact to an individual molecule is one of the biggest challenges in molecular optoelectronics. The mounting of individual chromophores on extended tripodal scaffolds enables both efficient electrical and mechanical decoupling of individual chromophores from metallic leads. Core-substituted naphthalene diimides fixed perpendicular to a gold substrate by a covalently attached extended tripod display high stability with well-defined and efficient electroluminescence down to the single-molecule level. The molecularly controlled spatial arrangement balances the electric conduction for electroluminescence and the insulation to avoid non-radiative carrier recombination, enabling the spectrally and spatially resolved electroluminescence of individual self-decoupled chromophores in a scanning tunneling microscope. Hot luminescence bands are even visible in single self-decoupled chromophores, documenting the mechanical decoupling between the vibrons of the chromophore and the substrate. |
| 106. |  | Jeongsu Pyeon, Soon Mo Park, Juri Kim, Jeong-Hwan Kim, Yong-Jin Yoon, Dong Ki Yoon, Hyoungsoo Kim Plasmonic metasurfaces of cellulose nanocrystal matrices with quadrants of aligned gold nanorods for photothermal anti-icing In: Nature Communications, vol. 14, no. 8096, 2023, (Cellulose nanocrystals are very attractive as a matrix material for plasmonic nanoparticles, but controlling particle orientation for patterning is challenging. Here, the authors prepare annular ring patterns with quadrants of aligned gold nanorods for photothermal applications.). @article{nokey,
title = {Plasmonic metasurfaces of cellulose nanocrystal matrices with quadrants of aligned gold nanorods for photothermal anti-icing},
author = {Jeongsu Pyeon and Soon Mo Park and Juri Kim and Jeong-Hwan Kim and Yong-Jin Yoon and Dong Ki Yoon and Hyoungsoo Kim },
url = {https://www.nature.com/articles/s41467-023-43511-9.pdf},
doi = {10.1038/s41467-023-43511-9},
year = {2023},
date = {2023-12-08},
urldate = {2023-12-08},
journal = {Nature Communications},
volume = {14},
number = {8096},
abstract = {Cellulose nanocrystals (CNCs) are intriguing as a matrix for plasmonic metasurfaces made of gold nanorods (GNRs) because of their distinctive properties, including renewability, biodegradability, non-toxicity, and low cost. Nevertheless, it is very difficult to precisely regulate the positioning and orientation of CNCs on the substrate in a consistent pattern. In this study, CNCs and GNRs, which exhibit tunable optical and anti-icing capabilities, are employed to manufacture a uniform plasmonic metasurface using a drop-casting technique. Two physical phenomena—(i) spontaneous and rapid self-dewetting and (ii) evaporation-induced self-assembly—are used to accomplish this. Additionally, we improve the CNC-GNR ink composition and determine the crucial coating parameters necessary to balance the two physical mechanisms in order to produce thin films without coffee rings. The final homogeneous CNC-GNR film has consistent annular ring patterns with plasmonic quadrant hues that are properly aligned, which enhances plasmonic photothermal effects. The CNC-GNR multi-array platform offers above-zero temperatures on a substrate that is subcooled below the freezing point. The current study presents a physicochemical approach for functional nanomaterial-based CNC control.},
note = {Cellulose nanocrystals are very attractive as a matrix material for plasmonic nanoparticles, but controlling particle orientation for patterning is challenging. Here, the authors prepare annular ring patterns with quadrants of aligned gold nanorods for photothermal applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cellulose nanocrystals (CNCs) are intriguing as a matrix for plasmonic metasurfaces made of gold nanorods (GNRs) because of their distinctive properties, including renewability, biodegradability, non-toxicity, and low cost. Nevertheless, it is very difficult to precisely regulate the positioning and orientation of CNCs on the substrate in a consistent pattern. In this study, CNCs and GNRs, which exhibit tunable optical and anti-icing capabilities, are employed to manufacture a uniform plasmonic metasurface using a drop-casting technique. Two physical phenomena—(i) spontaneous and rapid self-dewetting and (ii) evaporation-induced self-assembly—are used to accomplish this. Additionally, we improve the CNC-GNR ink composition and determine the crucial coating parameters necessary to balance the two physical mechanisms in order to produce thin films without coffee rings. The final homogeneous CNC-GNR film has consistent annular ring patterns with plasmonic quadrant hues that are properly aligned, which enhances plasmonic photothermal effects. The CNC-GNR multi-array platform offers above-zero temperatures on a substrate that is subcooled below the freezing point. The current study presents a physicochemical approach for functional nanomaterial-based CNC control. |
| 105. |  | Ziqing Zhang, Jinrun Dong, Yibo Yang, Yuan Zhou, Yuang Chen, Yang Xu, Jiandong Feng Direct probing of single-molecule chemiluminescent reaction dynamics under catalytic conditions in solution In: Nature Communications, vol. 14, no. 7993, 2023, (Research into the dynamics of chemical reactions at the single-molecule level is a pivotal undertaking. Here, the authors present a direct investigation of the chemiluminescent reaction dynamics of single molecules in solution, providing spatiotemporally resolved insights into chemical reactions.). @article{nokey,
title = {Direct probing of single-molecule chemiluminescent reaction dynamics under catalytic conditions in solution},
author = {Ziqing Zhang and Jinrun Dong and Yibo Yang and Yuan Zhou and Yuang Chen and Yang Xu and Jiandong Feng},
url = {https://www.nature.com/articles/s41467-023-43640-1.pdf},
doi = {10.1038/s41467-023-43640-1},
year = {2023},
date = {2023-12-02},
urldate = {2023-12-02},
journal = {Nature Communications},
volume = {14},
number = {7993},
abstract = {Chemical reaction kinetics can be evaluated by probing dynamic changes of chemical substrates or physical phenomena accompanied during the reaction process. Chemiluminescence, a light emitting exoenergetic process, involves random reaction positions and kinetics in solution that are typically characterized by ensemble measurements with nonnegligible average effects. Chemiluminescent reaction dynamics at the single-molecule level remains elusive. Here we report direct imaging of single-molecule chemiluminescent reactions in solution and probing of their reaction dynamics under catalytic conditions. Double-substrate Michaelis–Menten type of catalytic kinetics is found to govern the single-molecule reaction dynamics in solution, and a heterogeneity is found among different catalyst particles and different catalytic sites on a single particle. We further show that single-molecule chemiluminescence imaging can be used to evaluate the thermodynamics of the catalytic system, resolving activation energy at the single-particle level. Our work provides fundamental insights into chemiluminescent reactions and offers an efficient approach for evaluating catalysts.},
note = {Research into the dynamics of chemical reactions at the single-molecule level is a pivotal undertaking. Here, the authors present a direct investigation of the chemiluminescent reaction dynamics of single molecules in solution, providing spatiotemporally resolved insights into chemical reactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chemical reaction kinetics can be evaluated by probing dynamic changes of chemical substrates or physical phenomena accompanied during the reaction process. Chemiluminescence, a light emitting exoenergetic process, involves random reaction positions and kinetics in solution that are typically characterized by ensemble measurements with nonnegligible average effects. Chemiluminescent reaction dynamics at the single-molecule level remains elusive. Here we report direct imaging of single-molecule chemiluminescent reactions in solution and probing of their reaction dynamics under catalytic conditions. Double-substrate Michaelis–Menten type of catalytic kinetics is found to govern the single-molecule reaction dynamics in solution, and a heterogeneity is found among different catalyst particles and different catalytic sites on a single particle. We further show that single-molecule chemiluminescence imaging can be used to evaluate the thermodynamics of the catalytic system, resolving activation energy at the single-particle level. Our work provides fundamental insights into chemiluminescent reactions and offers an efficient approach for evaluating catalysts. |
| 104. |  | Shigeki Kawai, Orlando J. Silveira, Lauri Kurki, Zhangyu Yuan, Tomohiko Nishiuchi, Takuya Kodama, Kewei Sun, Oscar Custance, Jose L. Lado, Takashi Kubo, Adam S. Foster Local probe-induced structural isomerization in a one-dimensional molecular array In: Nature Communications, vol. 14, no. 7741, 2023, (Achieving unit-by-unit isomerization within a molecular array poses a significant challenge in chemistry. Here, the authors demonstrate tip-induced stereoisomerization of dehydroazulene and diradical units in three-dimensional organometallic compounds on Ag(111).). @article{nokey,
title = {Local probe-induced structural isomerization in a one-dimensional molecular array},
author = {Shigeki Kawai and Orlando J. Silveira and Lauri Kurki and Zhangyu Yuan and Tomohiko Nishiuchi and Takuya Kodama and Kewei Sun and Oscar Custance and Jose L. Lado and Takashi Kubo and Adam S. Foster },
url = {https://www.nature.com/articles/s41467-023-43659-4.pdf},
doi = {10.1038/s41467-023-43659-4},
year = {2023},
date = {2023-11-25},
urldate = {2023-11-25},
journal = {Nature Communications},
volume = {14},
number = {7741},
abstract = {Synthesis of one-dimensional molecular arrays with tailored stereoisomers is challenging yet has great potential for application in molecular opto-, electronic- and magnetic-devices, where the local array structure plays a decisive role in the functional properties. Here, we demonstrate the construction and characterization of dehydroazulene isomer and diradical units in three-dimensional organometallic compounds on Ag(111) with a combination of low-temperature scanning tunneling microscopy and density functional theory calculations. Tip-induced voltage pulses firstly result in the formation of a diradical species via successive homolytic fission of two C-Br bonds in the naphthyl groups, which are subsequently transformed into chiral dehydroazulene moieties. The delicate balance of the reaction rates among the diradical and two stereoisomers, arising from an in-line configuration of tip and molecular unit, allows directional azulene-to-azulene and azulene-to-diradical local probe structural isomerization in a controlled manner. Furthermore, our theoretical calculations suggest that the diradical moiety hosts an open-shell singlet with antiferromagnetic coupling between the unpaired electrons, which can undergo an inelastic spin transition of 91 meV to the ferromagnetically coupled triplet state.},
note = {Achieving unit-by-unit isomerization within a molecular array poses a significant challenge in chemistry. Here, the authors demonstrate tip-induced stereoisomerization of dehydroazulene and diradical units in three-dimensional organometallic compounds on Ag(111).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Synthesis of one-dimensional molecular arrays with tailored stereoisomers is challenging yet has great potential for application in molecular opto-, electronic- and magnetic-devices, where the local array structure plays a decisive role in the functional properties. Here, we demonstrate the construction and characterization of dehydroazulene isomer and diradical units in three-dimensional organometallic compounds on Ag(111) with a combination of low-temperature scanning tunneling microscopy and density functional theory calculations. Tip-induced voltage pulses firstly result in the formation of a diradical species via successive homolytic fission of two C-Br bonds in the naphthyl groups, which are subsequently transformed into chiral dehydroazulene moieties. The delicate balance of the reaction rates among the diradical and two stereoisomers, arising from an in-line configuration of tip and molecular unit, allows directional azulene-to-azulene and azulene-to-diradical local probe structural isomerization in a controlled manner. Furthermore, our theoretical calculations suggest that the diradical moiety hosts an open-shell singlet with antiferromagnetic coupling between the unpaired electrons, which can undergo an inelastic spin transition of 91 meV to the ferromagnetically coupled triplet state. |
| 103. |  | Peihui Li, Songjun Hou, Qingqing Wu, Yijian Chen, Boyu Wang, Haiyang Ren, Jinying Wang, Zhaoyi Zhai, Zhongbo Yu, Colin J. Lambert, Chuancheng Jia, Xuefeng Guo The role of halogens in Au–S bond cleavage for energy-differentiated catalysis at the single-bond limit In: Nature Communications, vol. 14, no. 7695, 2023, (Investigation of the reaction process at the single-bond interface is key to understanding the catalytic reaction mechanism. Here, the authors develop a STM-BJ method to monitor the catalytic process from the perspective of single-bond energy.). @article{nokey,
title = {The role of halogens in Au–S bond cleavage for energy-differentiated catalysis at the single-bond limit},
author = {Peihui Li and Songjun Hou and Qingqing Wu and Yijian Chen and Boyu Wang and Haiyang Ren and Jinying Wang and Zhaoyi Zhai and Zhongbo Yu and Colin J. Lambert and Chuancheng Jia and Xuefeng Guo },
url = {https://www.nature.com/articles/s41467-023-43639-8.pdf},
doi = {10.1038/s41467-023-43639-8},
year = {2023},
date = {2023-11-24},
urldate = {2023-11-24},
journal = {Nature Communications},
volume = {14},
number = {7695},
abstract = {The transformation from one compound to another involves the breaking and formation of chemical bonds at the single-bond level, especially during catalytic reactions that are of great significance in broad fields such as energy conversion, environmental science, life science and chemical synthesis. The study of the reaction process at the single-bond limit is the key to understanding the catalytic reaction mechanism and further rationally designing catalysts. Here, we develop a method to monitor the catalytic process from the perspective of the single-bond energy using high-resolution scanning tunneling microscopy single-molecule junctions. Experimental and theoretical studies consistently reveal that the attack of a halogen atom on an Au atom can reduce the breaking energy of Au−S bonds, thereby accelerating the bond cleavage reaction and shortening the plateau length during the single-molecule junction breaking. Furthermore, the distinction in catalytic activity between different halogen atoms can be compared as well. This study establishes the intrinsic relationship among the reaction activation energy, the chemical bond breaking energy and the single-molecule junction breaking process, strengthening our mastery of catalytic reactions towards precise chemistry.},
note = {Investigation of the reaction process at the single-bond interface is key to understanding the catalytic reaction mechanism. Here, the authors develop a STM-BJ method to monitor the catalytic process from the perspective of single-bond energy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The transformation from one compound to another involves the breaking and formation of chemical bonds at the single-bond level, especially during catalytic reactions that are of great significance in broad fields such as energy conversion, environmental science, life science and chemical synthesis. The study of the reaction process at the single-bond limit is the key to understanding the catalytic reaction mechanism and further rationally designing catalysts. Here, we develop a method to monitor the catalytic process from the perspective of the single-bond energy using high-resolution scanning tunneling microscopy single-molecule junctions. Experimental and theoretical studies consistently reveal that the attack of a halogen atom on an Au atom can reduce the breaking energy of Au−S bonds, thereby accelerating the bond cleavage reaction and shortening the plateau length during the single-molecule junction breaking. Furthermore, the distinction in catalytic activity between different halogen atoms can be compared as well. This study establishes the intrinsic relationship among the reaction activation energy, the chemical bond breaking energy and the single-molecule junction breaking process, strengthening our mastery of catalytic reactions towards precise chemistry. |
| 102. |  | Jacques Doumani, Minhan Lou, Oliver Dewey, Nina Hong, Jichao Fan, Andrey Baydin, Keshav Zahn, Yohei Yomogida, Kazuhiro Yanagi, Matteo Pasquali, Riichiro Saito, Junichiro Kono, Weilu Gao Engineering chirality at wafer scale with ordered carbon nanotube architectures In: Nature Communications, vol. 14, no. 7380, 2023, (Methods for generating macroscopic chiral matter struggle with limited scalability. Here, the authors show two vacuum filtration methods - twist stacking and mechanical rotation - to align carbon nanotubes into chiral structures at wafer scale with tunable circular dichroism.). @article{nokey,
title = {Engineering chirality at wafer scale with ordered carbon nanotube architectures},
author = {Jacques Doumani and Minhan Lou and Oliver Dewey and Nina Hong and Jichao Fan and Andrey Baydin and Keshav Zahn and Yohei Yomogida and Kazuhiro Yanagi and Matteo Pasquali and Riichiro Saito and Junichiro Kono and Weilu Gao },
url = {https://www.nature.com/articles/s41467-023-43199-x.pdf},
doi = {10.1038/s41467-023-43199-x},
year = {2023},
date = {2023-11-15},
urldate = {2023-11-15},
journal = {Nature Communications},
volume = {14},
number = {7380},
abstract = {Creating artificial matter with controllable chirality in a simple and scalable manner brings new opportunities to diverse areas. Here we show two such methods based on controlled vacuum filtration - twist stacking and mechanical rotation - for fabricating wafer-scale chiral architectures of ordered carbon nanotubes (CNTs) with tunable and large circular dichroism (CD). By controlling the stacking angle and handedness in the twist-stacking approach, we maximize the CD response and achieve a high deep-ultraviolet ellipticity of 40 ± 1 mdeg nm−1. Our theoretical simulations using the transfer matrix method reproduce the experimentally observed CD spectra and further predict that an optimized film of twist-stacked CNTs can exhibit an ellipticity as high as 150 mdeg nm−1, corresponding to a g factor of 0.22. Furthermore, the mechanical rotation method not only accelerates the fabrication of twisted structures but also produces both chiralities simultaneously in a single sample, in a single run, and in a controllable manner. The created wafer-scale objects represent an alternative type of synthetic chiral matter consisting of ordered quantum wires whose macroscopic properties are governed by nanoscopic electronic signatures and can be used to explore chiral phenomena and develop chiral photonic and optoelectronic devices.},
note = {Methods for generating macroscopic chiral matter struggle with limited scalability. Here, the authors show two vacuum filtration methods - twist stacking and mechanical rotation - to align carbon nanotubes into chiral structures at wafer scale with tunable circular dichroism.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Creating artificial matter with controllable chirality in a simple and scalable manner brings new opportunities to diverse areas. Here we show two such methods based on controlled vacuum filtration - twist stacking and mechanical rotation - for fabricating wafer-scale chiral architectures of ordered carbon nanotubes (CNTs) with tunable and large circular dichroism (CD). By controlling the stacking angle and handedness in the twist-stacking approach, we maximize the CD response and achieve a high deep-ultraviolet ellipticity of 40 ± 1 mdeg nm−1. Our theoretical simulations using the transfer matrix method reproduce the experimentally observed CD spectra and further predict that an optimized film of twist-stacked CNTs can exhibit an ellipticity as high as 150 mdeg nm−1, corresponding to a g factor of 0.22. Furthermore, the mechanical rotation method not only accelerates the fabrication of twisted structures but also produces both chiralities simultaneously in a single sample, in a single run, and in a controllable manner. The created wafer-scale objects represent an alternative type of synthetic chiral matter consisting of ordered quantum wires whose macroscopic properties are governed by nanoscopic electronic signatures and can be used to explore chiral phenomena and develop chiral photonic and optoelectronic devices. |
| 101. |  | Francis Schuknecht, Karol Kołątaj, Michael Steinberger, Tim Liedl, Theobald Lohmueller Accessible hotspots for single-protein SERS in DNA-origami assembled gold nanorod dimers with tip-to-tip alignment In: Nature Communications, vol. 14, no. 7192, 2023, (The identification of individual proteins is highly desirable in diagnostics. Here, the authors report on DNA-origami assembled dimers of gold nanorod with accessible hotspots to capture and identify single proteins from solution by SERS.). @article{nokey,
title = {Accessible hotspots for single-protein SERS in DNA-origami assembled gold nanorod dimers with tip-to-tip alignment},
author = {Francis Schuknecht and Karol Kołątaj and Michael Steinberger and Tim Liedl and Theobald Lohmueller },
url = {https://www.nature.com/articles/s41467-023-42943-7.pdf},
doi = {10.1038/s41467-023-42943-7},
year = {2023},
date = {2023-11-08},
urldate = {2023-11-08},
journal = {Nature Communications},
volume = {14},
number = {7192},
abstract = {The label-free identification of individual proteins from liquid samples by surface-enhanced Raman scattering (SERS) spectroscopy is a highly desirable goal in biomedical diagnostics. However, the small Raman scattering cross-section of most (bio-)molecules requires a means to strongly amplify their Raman signal for successful measurement, especially for single molecules. This amplification can be achieved in a plasmonic hotspot that forms between two adjacent gold nanospheres. However, the small (≈1−2 nm) gaps typically required for single-molecule measurements are not accessible for most proteins. A useful strategy would thus involve dimer structures with gaps large enough to accommodate single proteins, whilst providing sufficient field enhancement for single-molecule SERS. Here, we report on using a DNA origami scaffold for tip-to-tip alignment of gold nanorods with an average gap size of 8 nm. The gaps are accessible to streptavidin and thrombin, which are captured at the plasmonic hotspot by specific anchoring sites on the origami template. The field enhancement achieved for the nanorod dimers is sufficient for single-protein SERS spectroscopy with sub-second integration times. This design for SERS probes composed of DNA origami with accessible hotspots promotes future use for single-molecule biodiagnostics in the near-infrared range.},
note = {The identification of individual proteins is highly desirable in diagnostics. Here, the authors report on DNA-origami assembled dimers of gold nanorod with accessible hotspots to capture and identify single proteins from solution by SERS.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The label-free identification of individual proteins from liquid samples by surface-enhanced Raman scattering (SERS) spectroscopy is a highly desirable goal in biomedical diagnostics. However, the small Raman scattering cross-section of most (bio-)molecules requires a means to strongly amplify their Raman signal for successful measurement, especially for single molecules. This amplification can be achieved in a plasmonic hotspot that forms between two adjacent gold nanospheres. However, the small (≈1−2 nm) gaps typically required for single-molecule measurements are not accessible for most proteins. A useful strategy would thus involve dimer structures with gaps large enough to accommodate single proteins, whilst providing sufficient field enhancement for single-molecule SERS. Here, we report on using a DNA origami scaffold for tip-to-tip alignment of gold nanorods with an average gap size of 8 nm. The gaps are accessible to streptavidin and thrombin, which are captured at the plasmonic hotspot by specific anchoring sites on the origami template. The field enhancement achieved for the nanorod dimers is sufficient for single-protein SERS spectroscopy with sub-second integration times. This design for SERS probes composed of DNA origami with accessible hotspots promotes future use for single-molecule biodiagnostics in the near-infrared range. |
| 100. |  | Vivien Walter, Dadi Bi, Ali Salehi-Reyhani, Yansha Deng Real-time signal processing via chemical reactions for a microfluidic molecular communication system In: Nature Communications, vol. 14, no. 7188, 2023, (The use of electronic devices to process electrical signals in molecular communications can hardly realize its potential for various applications. Here, the authors report on chemical concentration signal processing in real time and digital signal transmission over distances.). @article{nokey,
title = {Real-time signal processing via chemical reactions for a microfluidic molecular communication system},
author = {Vivien Walter and Dadi Bi and Ali Salehi-Reyhani and Yansha Deng },
url = {https://www.nature.com/articles/s41467-023-42885-0.pdf},
doi = {10.1038/s41467-023-42885-0},
year = {2023},
date = {2023-11-08},
urldate = {2023-11-08},
journal = {Nature Communications},
volume = {14},
number = {7188},
abstract = {Signal processing over the molecular domain is critical for analysing, modifying, and synthesising chemical signals in molecular communication systems. However, the lack of chemical signal processing blocks and the wide use of electronic devices to process electrical signals in existing molecular communication platforms can hardly meet the biocompatible, non-invasive, and size-miniaturised requirements of applications in various fields, e.g., medicine, biology, and environment sciences. To tackle this, here we design and construct a liquid-based microfluidic molecular communication platform for performing chemical concentration signal processing and digital signal transmission over distances. By specifically designing chemical reactions and microfluidic geometry, the transmitter of our platform is capable of shaping the emitted signals, and the receiver is able to threshold, amplify, and detect the chemical signals after propagation. By encoding bit information into the concentration of sodium hydroxide, we demonstrate that our platform can achieve molecular signal modulation and demodulation functionalities, and reliably transmit text messages over long distances. This platform is further optimised to maximise data rate while minimising communication error. The presented methodology for real-time chemical signal processing can enable the implementation of signal processing units in biological settings and then unleash its potential for interdisciplinary applications.},
note = {The use of electronic devices to process electrical signals in molecular communications can hardly realize its potential for various applications. Here, the authors report on chemical concentration signal processing in real time and digital signal transmission over distances.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Signal processing over the molecular domain is critical for analysing, modifying, and synthesising chemical signals in molecular communication systems. However, the lack of chemical signal processing blocks and the wide use of electronic devices to process electrical signals in existing molecular communication platforms can hardly meet the biocompatible, non-invasive, and size-miniaturised requirements of applications in various fields, e.g., medicine, biology, and environment sciences. To tackle this, here we design and construct a liquid-based microfluidic molecular communication platform for performing chemical concentration signal processing and digital signal transmission over distances. By specifically designing chemical reactions and microfluidic geometry, the transmitter of our platform is capable of shaping the emitted signals, and the receiver is able to threshold, amplify, and detect the chemical signals after propagation. By encoding bit information into the concentration of sodium hydroxide, we demonstrate that our platform can achieve molecular signal modulation and demodulation functionalities, and reliably transmit text messages over long distances. This platform is further optimised to maximise data rate while minimising communication error. The presented methodology for real-time chemical signal processing can enable the implementation of signal processing units in biological settings and then unleash its potential for interdisciplinary applications. |
| 99. | 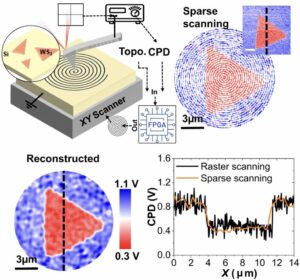 | Marti Checa, Addis S. Fuhr, Changhyo Sun, Rama Vasudevan, Maxim Ziatdinov, Ilia Ivanov, Seok Joon Yun, Kai Xiao, Alp Sehirlioglu, Yunseok Kim, Pankaj Sharma, Kyle P. Kelley, Neus Domingo, Stephen Jesse, Liam Collins High-speed mapping of surface charge dynamics using sparse scanning Kelvin probe force microscopy In: Nature Communications, vol. 14, no. 7196, 2023, (Dynamic mapping of charge motion across multiple length- and timescales is essential for understanding a variety of phenomena. Here, the authors introduce sparse scanning KPFM, which enables fast nanoscale charge mapping at 3 frames per second to track ion migration.). @article{nokey,
title = {High-speed mapping of surface charge dynamics using sparse scanning Kelvin probe force microscopy},
author = {Marti Checa and Addis S. Fuhr and Changhyo Sun and Rama Vasudevan and Maxim Ziatdinov and Ilia Ivanov and Seok Joon Yun and Kai Xiao and Alp Sehirlioglu and Yunseok Kim and Pankaj Sharma and Kyle P. Kelley and Neus Domingo and Stephen Jesse and Liam Collins },
url = {https://www.nature.com/articles/s41467-023-42583-x.pdf},
doi = {10.1038/s41467-023-42583-x},
year = {2023},
date = {2023-11-08},
urldate = {2023-11-08},
journal = {Nature Communications},
volume = {14},
number = {7196},
abstract = {Unraveling local dynamic charge processes is vital for progress in diverse fields, from microelectronics to energy storage. This relies on the ability to map charge carrier motion across multiple length- and timescales and understanding how these processes interact with the inherent material heterogeneities. Towards addressing this challenge, we introduce high-speed sparse scanning Kelvin probe force microscopy, which combines sparse scanning and image reconstruction. This approach is shown to enable sub-second imaging (>3 frames per second) of nanoscale charge dynamics, representing several orders of magnitude improvement over traditional Kelvin probe force microscopy imaging rates. Bridging this improved spatiotemporal resolution with macroscale device measurements, we successfully visualize electrochemically mediated diffusion of mobile surface ions on a LaAlO3/SrTiO3 planar device. Such processes are known to impact band-alignment and charge-transfer dynamics at these heterointerfaces. Furthermore, we monitor the diffusion of oxygen vacancies at the single grain level in polycrystalline TiO2. Through temperature-dependent measurements, we identify a charge diffusion activation energy of 0.18 eV, in good agreement with previously reported values and confirmed by DFT calculations. Together, these findings highlight the effectiveness and versatility of our method in understanding ionic charge carrier motion in microelectronics or nanoscale material systems.},
note = {Dynamic mapping of charge motion across multiple length- and timescales is essential for understanding a variety of phenomena. Here, the authors introduce sparse scanning KPFM, which enables fast nanoscale charge mapping at 3 frames per second to track ion migration.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Unraveling local dynamic charge processes is vital for progress in diverse fields, from microelectronics to energy storage. This relies on the ability to map charge carrier motion across multiple length- and timescales and understanding how these processes interact with the inherent material heterogeneities. Towards addressing this challenge, we introduce high-speed sparse scanning Kelvin probe force microscopy, which combines sparse scanning and image reconstruction. This approach is shown to enable sub-second imaging (>3 frames per second) of nanoscale charge dynamics, representing several orders of magnitude improvement over traditional Kelvin probe force microscopy imaging rates. Bridging this improved spatiotemporal resolution with macroscale device measurements, we successfully visualize electrochemically mediated diffusion of mobile surface ions on a LaAlO3/SrTiO3 planar device. Such processes are known to impact band-alignment and charge-transfer dynamics at these heterointerfaces. Furthermore, we monitor the diffusion of oxygen vacancies at the single grain level in polycrystalline TiO2. Through temperature-dependent measurements, we identify a charge diffusion activation energy of 0.18 eV, in good agreement with previously reported values and confirmed by DFT calculations. Together, these findings highlight the effectiveness and versatility of our method in understanding ionic charge carrier motion in microelectronics or nanoscale material systems. |
| 98. |  | Zhiwen Jiang, Carine Clavaguéra, Changjiang Hu, Sergey A. Denisov, Shuning Shen, Feng Hu, Jun Ma, Mehran Mostafavi Direct time-resolved observation of surface-bound carbon dioxide radical anions on metallic nanocatalysts In: Nature Communications, vol. 14, no. 7116, 2023, (Understanding the activity and selectivity of metal catalysts requires elucidating the dynamics of CO2•− radicals bound to the surface. Here, the authors use pulse radiolysis to directly observe the stabilization process of CO2•− radicals at nanoscale metallic sites from nanoseconds to seconds.). @article{nokey,
title = {Direct time-resolved observation of surface-bound carbon dioxide radical anions on metallic nanocatalysts},
author = {Zhiwen Jiang and Carine Clavaguéra and Changjiang Hu and Sergey A. Denisov and Shuning Shen and Feng Hu and Jun Ma and Mehran Mostafavi },
url = {https://www.nature.com/articles/s41467-023-42936-6.pdf},
doi = {10.1038/s41467-023-42936-6},
year = {2023},
date = {2023-11-06},
urldate = {2023-11-06},
journal = {Nature Communications},
volume = {14},
number = {7116},
abstract = {Time-resolved identification of surface-bound intermediates on metallic nanocatalysts is imperative to develop an accurate understanding of the elementary steps of CO2 reduction. Direct observation on initial electron transfer to CO2 to form surface-bound CO2•− radicals is lacking due to the technical challenges. Here, we use picosecond pulse radiolysis to generate CO2•− via aqueous electron attachment and observe the stabilization processes toward well-defined nanoscale metallic sites. The time-resolved method combined with molecular simulations identifies surface-bound intermediates with characteristic transient absorption bands and distinct kinetics from nanosecond to the second timescale for three typical metallic nanocatalysts: Cu, Au, and Ni. The interfacial interactions are further investigated by varying the important factors, such as catalyst size and the presence of cation in the electrolyte. This work highlights fundamental ultrafast spectroscopy to clarify the critical initial step in the CO2 catalytic reduction mechanism.},
note = {Understanding the activity and selectivity of metal catalysts requires elucidating the dynamics of CO2•− radicals bound to the surface. Here, the authors use pulse radiolysis to directly observe the stabilization process of CO2•− radicals at nanoscale metallic sites from nanoseconds to seconds.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Time-resolved identification of surface-bound intermediates on metallic nanocatalysts is imperative to develop an accurate understanding of the elementary steps of CO2 reduction. Direct observation on initial electron transfer to CO2 to form surface-bound CO2•− radicals is lacking due to the technical challenges. Here, we use picosecond pulse radiolysis to generate CO2•− via aqueous electron attachment and observe the stabilization processes toward well-defined nanoscale metallic sites. The time-resolved method combined with molecular simulations identifies surface-bound intermediates with characteristic transient absorption bands and distinct kinetics from nanosecond to the second timescale for three typical metallic nanocatalysts: Cu, Au, and Ni. The interfacial interactions are further investigated by varying the important factors, such as catalyst size and the presence of cation in the electrolyte. This work highlights fundamental ultrafast spectroscopy to clarify the critical initial step in the CO2 catalytic reduction mechanism. |
| 97. |  | Bowen Sui, Youliang Zhu, Xuemei Jiang, Yifan Wang, Niboqia Zhang, Zhongyuan Lu, Bai Yang, Yunfeng Li Recastable assemblies of carbon dots into mechanically robust macroscopic materials In: Nature Communications, vol. 14, no. 6782, 2023, (The assembly of nanoparticles into macroscopic materials forms the basis for various advanced material applications. Here, the authors develop macroscopic materials that are both recastable and mechanically robust, despite being composed purely of carbon dots.). @article{nokey,
title = {Recastable assemblies of carbon dots into mechanically robust macroscopic materials},
author = {Bowen Sui and Youliang Zhu and Xuemei Jiang and Yifan Wang and Niboqia Zhang and Zhongyuan Lu and Bai Yang and Yunfeng Li },
url = {https://www.nature.com/articles/s41467-023-42516-8.pdf},
doi = {10.1038/s41467-023-42516-8},
year = {2023},
date = {2023-10-24},
urldate = {2023-10-24},
journal = {Nature Communications},
volume = {14},
number = {6782},
abstract = {Assembly of nanoparticles into macroscopic materials with mechanical robustness, green processability, and recastable ability is an important and challenging task in materials science and nanotechnology. As an emerging nanoparticle with superior properties, macroscopic materials assembled from carbon dots will inherit their properties and further offer collective properties; however, macroscopic materials assembled from carbon dots solely remain unexplored. Here we report macroscopic films assembled from carbon dots modified by ureido pyrimidinone. These films show tunable fluorescence inherited from carbon dots. More importantly, these films exhibit collective properties including self-healing, re-castability, and superior mechanical properties, with Young’s modulus over 490 MPa and breaking strength over 30 MPa. The macroscopic films maintain original mechanical properties after several cycles of recasting. Through scratch healing and welding experiments, these films show good self-healing properties under mild conditions. Moreover, the molecular dynamics simulation reveals that the interplay of interparticle and intraparticle hydrogen bonding controls mechanical properties of macroscopic films. Notably, these films are processed into diverse shapes by an eco-friendly hydrosetting method. The methodology and results in this work shed light on the exploration of functional macroscopic materials assembled from nanoparticles and will accelerate innovative developments of nanomaterials in practical applications.},
note = {The assembly of nanoparticles into macroscopic materials forms the basis for various advanced material applications. Here, the authors develop macroscopic materials that are both recastable and mechanically robust, despite being composed purely of carbon dots.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Assembly of nanoparticles into macroscopic materials with mechanical robustness, green processability, and recastable ability is an important and challenging task in materials science and nanotechnology. As an emerging nanoparticle with superior properties, macroscopic materials assembled from carbon dots will inherit their properties and further offer collective properties; however, macroscopic materials assembled from carbon dots solely remain unexplored. Here we report macroscopic films assembled from carbon dots modified by ureido pyrimidinone. These films show tunable fluorescence inherited from carbon dots. More importantly, these films exhibit collective properties including self-healing, re-castability, and superior mechanical properties, with Young’s modulus over 490 MPa and breaking strength over 30 MPa. The macroscopic films maintain original mechanical properties after several cycles of recasting. Through scratch healing and welding experiments, these films show good self-healing properties under mild conditions. Moreover, the molecular dynamics simulation reveals that the interplay of interparticle and intraparticle hydrogen bonding controls mechanical properties of macroscopic films. Notably, these films are processed into diverse shapes by an eco-friendly hydrosetting method. The methodology and results in this work shed light on the exploration of functional macroscopic materials assembled from nanoparticles and will accelerate innovative developments of nanomaterials in practical applications. |
| 96. |  | Alexander M. Bergmann, Jonathan Bauermann, Giacomo Bartolucci, Carsten Donau,
Michele Stasi, Anna-Lena Holtmannspötter, Frank Jülicher, Christoph A. Weber, Job Boekhoven Liquid spherical shells are a non-equilibrium steady state of active droplets In: Nature Communications, vol. 14, no. 6552, 2023, (Dissipative structures are governed by non-equilibrium thermodynamics. Here, the authors describe a size-dependent transition from active droplets to active spherical shells—a dissipative structure that arises from reaction diffusion gradients. ). @article{nokey,
title = {Liquid spherical shells are a non-equilibrium steady state of active droplets},
author = {Alexander M. Bergmann and Jonathan Bauermann and Giacomo Bartolucci and Carsten Donau and
Michele Stasi and Anna-Lena Holtmannspötter and Frank Jülicher and Christoph A. Weber and Job Boekhoven},
url = {https://www.nature.com/articles/s41467-023-42344-w.pdf},
doi = {10.1038/s41467-023-42344-w},
year = {2023},
date = {2023-10-17},
urldate = {2023-10-17},
journal = {Nature Communications},
volume = {14},
number = {6552},
abstract = {Liquid-liquid phase separation yields spherical droplets that eventually coarsen to one large, stable droplet governed by the principle of minimal free energy. In chemically fueled phase separation, the formation of phase-separating molecules is coupled to a fuel-driven, non-equilibrium reaction cycle. It thus yields dissipative structures sustained by a continuous fuel conversion. Such dissipative structures are ubiquitous in biology but are poorly understood as they are governed by non-equilibrium thermodynamics. Here, we bridge the gap between passive, close-to-equilibrium, and active, dissipative structures with chemically fueled phase separation. We observe that spherical, active droplets can undergo a morphological transition into a liquid, spherical shell. We demonstrate that the mechanism is related to gradients of short-lived droplet material. We characterize how far out of equilibrium the spherical shell state is and the chemical power necessary to sustain it. Our work suggests alternative avenues for assembling complex stable morphologies, which might already be exploited to form membraneless organelles by cells.},
note = {Dissipative structures are governed by non-equilibrium thermodynamics. Here, the authors describe a size-dependent transition from active droplets to active spherical shells—a dissipative structure that arises from reaction diffusion gradients. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Liquid-liquid phase separation yields spherical droplets that eventually coarsen to one large, stable droplet governed by the principle of minimal free energy. In chemically fueled phase separation, the formation of phase-separating molecules is coupled to a fuel-driven, non-equilibrium reaction cycle. It thus yields dissipative structures sustained by a continuous fuel conversion. Such dissipative structures are ubiquitous in biology but are poorly understood as they are governed by non-equilibrium thermodynamics. Here, we bridge the gap between passive, close-to-equilibrium, and active, dissipative structures with chemically fueled phase separation. We observe that spherical, active droplets can undergo a morphological transition into a liquid, spherical shell. We demonstrate that the mechanism is related to gradients of short-lived droplet material. We characterize how far out of equilibrium the spherical shell state is and the chemical power necessary to sustain it. Our work suggests alternative avenues for assembling complex stable morphologies, which might already be exploited to form membraneless organelles by cells. |
| 95. | 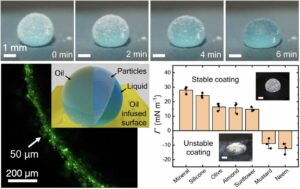 | Rutvik Lathia, Satchit Nagpal, Chandantaru Dey Modak, Satyarthi Mishra, Deepak Sharma, Bheema Sankar Reddy, Pavan Nukala, Ramray Bhat, Prosenjit Sen Tunable encapsulation of sessile droplets with solid and liquid shells In: Nature Communications, vol. 14, no. 6445, 2023, (Encapsulated liquids are important for several microreactor applications, including (bio)chemistry in confined spaces. Here, the authors report on oil-infused particle shells that allow control of shell thickness, stability, and permeability for applications in crystal growth and cell cultivation.). @article{nokey,
title = {Tunable encapsulation of sessile droplets with solid and liquid shells},
author = {Rutvik Lathia and Satchit Nagpal and Chandantaru Dey Modak and Satyarthi Mishra and Deepak Sharma and Bheema Sankar Reddy and Pavan Nukala and Ramray Bhat and Prosenjit Sen },
url = {https://www.nature.com/articles/s41467-023-41977-1.pdf},
doi = {10.1038/s41467-023-41977-1},
year = {2023},
date = {2023-10-13},
urldate = {2023-10-13},
journal = {Nature Communications},
volume = {14},
number = {6445},
abstract = {Droplet encapsulations using liquid or solid shells are of significant interest in microreactors, drug delivery, crystallization, and cell growth applications. Despite progress in droplet-related technologies, tuning micron-scale shell thickness over a large range of droplet sizes is still a major challenge. In this work, we report capillary force assisted cloaking using hydrophobic colloidal particles and liquid-infused surfaces. The technique produces uniform solid and liquid shell encapsulations over a broad range (5–200 μm shell thickness for droplet volume spanning over four orders of magnitude). Tunable liquid encapsulation is shown to reduce the evaporation rate of droplets by up to 200 times with a wide tunability in lifetime (1.5 h to 12 days). Further, we propose using the technique for single crystals and cell/spheroid culture platforms. Stimuli-responsive solid shells show hermetic encapsulation with tunable strength and dissolution time. Moreover, scalability, and versatility of the technique is demonstrated for on-chip applications.},
note = {Encapsulated liquids are important for several microreactor applications, including (bio)chemistry in confined spaces. Here, the authors report on oil-infused particle shells that allow control of shell thickness, stability, and permeability for applications in crystal growth and cell cultivation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Droplet encapsulations using liquid or solid shells are of significant interest in microreactors, drug delivery, crystallization, and cell growth applications. Despite progress in droplet-related technologies, tuning micron-scale shell thickness over a large range of droplet sizes is still a major challenge. In this work, we report capillary force assisted cloaking using hydrophobic colloidal particles and liquid-infused surfaces. The technique produces uniform solid and liquid shell encapsulations over a broad range (5–200 μm shell thickness for droplet volume spanning over four orders of magnitude). Tunable liquid encapsulation is shown to reduce the evaporation rate of droplets by up to 200 times with a wide tunability in lifetime (1.5 h to 12 days). Further, we propose using the technique for single crystals and cell/spheroid culture platforms. Stimuli-responsive solid shells show hermetic encapsulation with tunable strength and dissolution time. Moreover, scalability, and versatility of the technique is demonstrated for on-chip applications. |
| 94. | 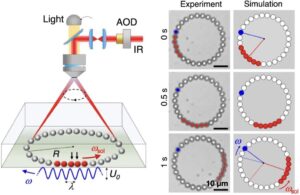 | Eric Cereceda-López, Alexander P. Antonov, Artem Ryabov, Philipp Maass, Pietro Tierno Overcrowding induces fast colloidal solitons in a slowly rotating potential landscape In: Nature Communications, vol. 14, no. 6448, 2023, (Solitons are peculiar waves propagating without changing their shape. Here, the authors show that colloidal particles in a rotating optical landscape create rapidly propagating solitons, formed by particle clusters through many-body interactions.). @article{nokey,
title = {Overcrowding induces fast colloidal solitons in a slowly rotating potential landscape},
author = {Eric Cereceda-López and Alexander P. Antonov and Artem Ryabov and Philipp Maass and Pietro Tierno },
url = {https://www.nature.com/articles/s41467-023-41989-x.pdf},
doi = {10.1038/s41467-023-41989-x},
year = {2023},
date = {2023-10-13},
urldate = {2023-10-13},
journal = {Nature Communications},
volume = {14},
number = {6448},
abstract = {Collective particle transport across periodic energy landscapes is ubiquitously present in many condensed matter systems spanning from vortices in high-temperature superconductors, frictional atomic sliding, driven skyrmions to biological and active matter. Here we report the emergence of fast solitons propagating against a rotating optical landscape. These experimentally observed solitons are stable cluster waves that originate from a coordinated particle exchange process which occurs when the number of trapped microparticles exceeds the number of potential wells. The size and speed of individual solitons rapidly increase with the particle diameter as predicted by theory and confirmed by numerical simulations. We show that when several solitons coexist, an effective repulsive interaction can stabilize their propagation along the periodic potential. Our experiments demonstrate a generic mechanism for cluster-mediated transport with potential applications to condensed matter systems on different length scales.},
note = {Solitons are peculiar waves propagating without changing their shape. Here, the authors show that colloidal particles in a rotating optical landscape create rapidly propagating solitons, formed by particle clusters through many-body interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Collective particle transport across periodic energy landscapes is ubiquitously present in many condensed matter systems spanning from vortices in high-temperature superconductors, frictional atomic sliding, driven skyrmions to biological and active matter. Here we report the emergence of fast solitons propagating against a rotating optical landscape. These experimentally observed solitons are stable cluster waves that originate from a coordinated particle exchange process which occurs when the number of trapped microparticles exceeds the number of potential wells. The size and speed of individual solitons rapidly increase with the particle diameter as predicted by theory and confirmed by numerical simulations. We show that when several solitons coexist, an effective repulsive interaction can stabilize their propagation along the periodic potential. Our experiments demonstrate a generic mechanism for cluster-mediated transport with potential applications to condensed matter systems on different length scales. |
| 93. |  | Deepak Karna, Eriko Mano, Jiahao Ji, Ibuki Kawamata, Yuki Suzuki, Hanbin Mao Chemo-mechanical forces modulate the topology dynamics of mesoscale DNA assemblies In: Nature Communications, vol. 14, no. 6459, 2023, (Understanding the topological arrangement and transition dynamics of mesoscale assemblies is complicated by their molecular complexity. Here, the authors use DNA origami nanosprings to show that mesoscale helical handedness is dictated by backbone torque rather than achiral orientation.). @article{nokey,
title = {Chemo-mechanical forces modulate the topology dynamics of mesoscale DNA assemblies},
author = {Deepak Karna and Eriko Mano and Jiahao Ji and Ibuki Kawamata and Yuki Suzuki and Hanbin Mao },
url = {https://www.nature.com/articles/s41467-023-41604-z.pdf},
doi = {10.1038/s41467-023-41604-z},
year = {2023},
date = {2023-10-13},
urldate = {2023-10-13},
journal = {Nature Communications},
volume = {14},
number = {6459},
abstract = {The intrinsic complexity of many mesoscale (10–100 nm) cellular machineries makes it challenging to elucidate their topological arrangement and transition dynamics. Here, we exploit DNA origami nanospring as a model system to demonstrate that tens of piconewton linear force can modulate higher-order conformation dynamics of mesoscale molecular assemblies. By switching between two chemical structures (i.e., duplex and tetraplex DNA) in the junctions of adjacent origami modules, the corresponding stretching or compressing chemo-mechanical stress reversibly flips the backbone orientations of the DNA nanosprings. Both coarse-grained molecular dynamics simulations and atomic force microscopy measurements reveal that such a backbone conformational switch does not alter the right-handed chirality of the nanospring helix. This result suggests that mesoscale helical handedness may be governed by the torque, rather than the achiral orientation, of nanospring backbones. It offers a topology-based caging/uncaging concept to present chemicals in response to environmental cues in solution.},
note = {Understanding the topological arrangement and transition dynamics of mesoscale assemblies is complicated by their molecular complexity. Here, the authors use DNA origami nanosprings to show that mesoscale helical handedness is dictated by backbone torque rather than achiral orientation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The intrinsic complexity of many mesoscale (10–100 nm) cellular machineries makes it challenging to elucidate their topological arrangement and transition dynamics. Here, we exploit DNA origami nanospring as a model system to demonstrate that tens of piconewton linear force can modulate higher-order conformation dynamics of mesoscale molecular assemblies. By switching between two chemical structures (i.e., duplex and tetraplex DNA) in the junctions of adjacent origami modules, the corresponding stretching or compressing chemo-mechanical stress reversibly flips the backbone orientations of the DNA nanosprings. Both coarse-grained molecular dynamics simulations and atomic force microscopy measurements reveal that such a backbone conformational switch does not alter the right-handed chirality of the nanospring helix. This result suggests that mesoscale helical handedness may be governed by the torque, rather than the achiral orientation, of nanospring backbones. It offers a topology-based caging/uncaging concept to present chemicals in response to environmental cues in solution. |
| 92. |  | Marloes H. Bistervels, Balázs Antalicz, Marko Kamp, Hinco Schoenmaker, Willem L. Noorduin Light-driven nucleation, growth, and patterning of biorelevant crystals using resonant near-infrared laser heating In: Nature Communications, vol. 14, no. 6350, 2023, (Natural biominerals deposit with precise spatial and temporal control, but such control is difficult to replicate artificially. Here, the authors start, steer, and stop the crystallization of biorelevant minerals by user-defined light patterns.). @article{nokey,
title = {Light-driven nucleation, growth, and patterning of biorelevant crystals using resonant near-infrared laser heating},
author = {Marloes H. Bistervels and Balázs Antalicz and Marko Kamp and Hinco Schoenmaker and Willem L. Noorduin },
url = {https://www.nature.com/articles/s41467-023-42126-4.pdf},
doi = {10.1038/s41467-023-42126-4},
year = {2023},
date = {2023-10-10},
urldate = {2023-10-10},
journal = {Nature Communications},
volume = {14},
number = {6350},
abstract = {Spatiotemporal control over crystal nucleation and growth is of fundamental interest for understanding how organisms assemble high-performance biominerals, and holds relevance for manufacturing of functional materials. Many methods have been developed towards static or global control, however gaining simultaneously dynamic and local control over crystallization remains challenging. Here, we show spatiotemporal control over crystallization of retrograde (inverse) soluble compounds induced by locally heating water using near-infrared (NIR) laser light. We modulate the NIR light intensity to start, steer, and stop crystallization of calcium carbonate and laser-write with micrometer precision. Tailoring the crystallization conditions overcomes the inherently stochastic crystallization behavior and enables positioning single crystals of vaterite, calcite, and aragonite. We demonstrate straightforward extension of these principles toward other biorelevant compounds by patterning barium-, strontium-, and calcium carbonate, as well as strontium sulfate and calcium phosphate. Since many important compounds exhibit retrograde solubility behavior, NIR-induced heating may enable light-controlled crystallization with precise spatiotemporal control.},
note = {Natural biominerals deposit with precise spatial and temporal control, but such control is difficult to replicate artificially. Here, the authors start, steer, and stop the crystallization of biorelevant minerals by user-defined light patterns.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Spatiotemporal control over crystal nucleation and growth is of fundamental interest for understanding how organisms assemble high-performance biominerals, and holds relevance for manufacturing of functional materials. Many methods have been developed towards static or global control, however gaining simultaneously dynamic and local control over crystallization remains challenging. Here, we show spatiotemporal control over crystallization of retrograde (inverse) soluble compounds induced by locally heating water using near-infrared (NIR) laser light. We modulate the NIR light intensity to start, steer, and stop crystallization of calcium carbonate and laser-write with micrometer precision. Tailoring the crystallization conditions overcomes the inherently stochastic crystallization behavior and enables positioning single crystals of vaterite, calcite, and aragonite. We demonstrate straightforward extension of these principles toward other biorelevant compounds by patterning barium-, strontium-, and calcium carbonate, as well as strontium sulfate and calcium phosphate. Since many important compounds exhibit retrograde solubility behavior, NIR-induced heating may enable light-controlled crystallization with precise spatiotemporal control. |
| 91. |  | Aritra Biswas, Nir Lemcoff, Ofir Shelonchik, Doron Yesodi, Elad Yehezkel, Ella Yonit Finestone, Alexander Upcher, Yossi Weizmann Photothermally heated colloidal synthesis of nanoparticles driven by silica-encapsulated plasmonic heat sources In: Nature Communications, vol. 14, no. 6355, 2023, (Photothermal solution heating with nanoscale heat sources is an efficient alternative to conventional heating methods. Here, the authors use silica-encapsulated gold nanoparticles to drive the colloidal synthesis of iron oxide, silver, and palladium nanoparticles at lower temperatures.). @article{nokey,
title = {Photothermally heated colloidal synthesis of nanoparticles driven by silica-encapsulated plasmonic heat sources},
author = {Aritra Biswas and Nir Lemcoff and Ofir Shelonchik and Doron Yesodi and Elad Yehezkel and Ella Yonit Finestone and Alexander Upcher and Yossi Weizmann },
url = {https://www.nature.com/articles/s41467-023-42167-9.pdf},
doi = {10.1038/s41467-023-42167-9},
year = {2023},
date = {2023-10-10},
urldate = {2023-10-10},
journal = {Nature Communications},
volume = {14},
number = {6355},
abstract = {Using photons to drive chemical reactions has become an increasingly important field of chemistry. Plasmonic materials can provide a means to introduce the energy necessary for nucleation and growth of nanoparticles by efficiently converting visible and infrared light to heat. Moreover, the formation of crystalline nanoparticles has yet to be included in the extensive list of plasmonic photothermal processes. Herein, we establish a light-assisted colloidal synthesis of iron oxide, silver, and palladium nanoparticles by utilizing silica-encapsulated gold bipyramids as plasmonic heat sources. Our work shows that the silica surface chemistry and localized thermal hotspot generated by the plasmonic nanoparticles play crucial roles in the formation mechanism, enabling nucleation and growth at temperatures considerably lower than conventional heating. Additionally, the photothermal method is extended to anisotropic geometries and can be applied to obtain intricate assemblies inaccessible otherwise. This study enables photothermally heated nanoparticle synthesis in solution through the plasmonic effect and demonstrates the potential of this methodology.},
note = {Photothermal solution heating with nanoscale heat sources is an efficient alternative to conventional heating methods. Here, the authors use silica-encapsulated gold nanoparticles to drive the colloidal synthesis of iron oxide, silver, and palladium nanoparticles at lower temperatures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Using photons to drive chemical reactions has become an increasingly important field of chemistry. Plasmonic materials can provide a means to introduce the energy necessary for nucleation and growth of nanoparticles by efficiently converting visible and infrared light to heat. Moreover, the formation of crystalline nanoparticles has yet to be included in the extensive list of plasmonic photothermal processes. Herein, we establish a light-assisted colloidal synthesis of iron oxide, silver, and palladium nanoparticles by utilizing silica-encapsulated gold bipyramids as plasmonic heat sources. Our work shows that the silica surface chemistry and localized thermal hotspot generated by the plasmonic nanoparticles play crucial roles in the formation mechanism, enabling nucleation and growth at temperatures considerably lower than conventional heating. Additionally, the photothermal method is extended to anisotropic geometries and can be applied to obtain intricate assemblies inaccessible otherwise. This study enables photothermally heated nanoparticle synthesis in solution through the plasmonic effect and demonstrates the potential of this methodology. |
| 90. |  | S. Furkan Ozturk, Deb Kumar Bhowmick, Yael Kapon, Yutao Sang, Anil Kumar, Yossi Paltiel, Ron Naaman, Dimitar D. Sasselov Chirality-induced avalanche magnetization of magnetite by an RNA precursor In: Nature Communications, vol. 14, no. 6351, 2023, (Homochirality, a key feature of life, has unknown origins. Magnetic mineral surfaces can act as chiral agents, but are only weakly magnetized by nature. Here, the authors report the uniform magnetization of magnetite by an RNA precursor that spreads across the surface like an avalanche.). @article{nokey,
title = {Chirality-induced avalanche magnetization of magnetite by an RNA precursor},
author = {S. Furkan Ozturk and Deb Kumar Bhowmick and Yael Kapon and Yutao Sang and Anil Kumar and Yossi Paltiel and Ron Naaman and Dimitar D. Sasselov },
url = {https://www.nature.com/articles/s41467-023-42130-8.pdf},
doi = {10.1038/s41467-023-42130-8},
year = {2023},
date = {2023-10-10},
urldate = {2023-10-10},
journal = {Nature Communications},
volume = {14},
number = {6351},
abstract = {Homochirality is a hallmark of life on Earth. To achieve and maintain homochirality within a prebiotic network, the presence of an environmental factor acting as a chiral agent and providing a persistent chiral bias to prebiotic chemistry is highly advantageous. Magnetized surfaces are prebiotically plausible chiral agents due to the chiral-induced spin selectivity (CISS) effect, and they were utilized to attain homochiral ribose-aminooxazoline (RAO), an RNA precursor. However, natural magnetic minerals are typically weakly magnetized, necessitating mechanisms to enhance their magnetization for their use as effective chiral agents. Here, we report the magnetization of magnetic surfaces by crystallizing enantiopure RAO, whereby chiral molecules induce a uniform surface magnetization due to the CISS effect, which spreads across the magnetic surface akin to an avalanche. Chirality-induced avalanche magnetization enables a feedback between chiral molecules and magnetic surfaces, which can amplify a weak magnetization and allow for highly efficient spin-selective processes on magnetic minerals.},
note = {Homochirality, a key feature of life, has unknown origins. Magnetic mineral surfaces can act as chiral agents, but are only weakly magnetized by nature. Here, the authors report the uniform magnetization of magnetite by an RNA precursor that spreads across the surface like an avalanche.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Homochirality is a hallmark of life on Earth. To achieve and maintain homochirality within a prebiotic network, the presence of an environmental factor acting as a chiral agent and providing a persistent chiral bias to prebiotic chemistry is highly advantageous. Magnetized surfaces are prebiotically plausible chiral agents due to the chiral-induced spin selectivity (CISS) effect, and they were utilized to attain homochiral ribose-aminooxazoline (RAO), an RNA precursor. However, natural magnetic minerals are typically weakly magnetized, necessitating mechanisms to enhance their magnetization for their use as effective chiral agents. Here, we report the magnetization of magnetic surfaces by crystallizing enantiopure RAO, whereby chiral molecules induce a uniform surface magnetization due to the CISS effect, which spreads across the magnetic surface akin to an avalanche. Chirality-induced avalanche magnetization enables a feedback between chiral molecules and magnetic surfaces, which can amplify a weak magnetization and allow for highly efficient spin-selective processes on magnetic minerals. |
| 89. |  | Lu Zhang, Wencai Yi, Junfang Li, Guoying Wei, Guangcheng Xi, Lanqun Mao Surfactant-free interfacial growth of graphdiyne hollow microspheres and the mechanistic origin of their SERS activity In: Nature Communications, vol. 14, no. 6318, 2023, (The 2D carbon allotrope graphdiyne possesses a direct band gap, high charge carrier mobility, and uniformly distributed pores. Here, the authors report the surfactant-free growth of graphdiyne hollow microspheres and investigate the origin of their SERS activity.). @article{nokey,
title = {Surfactant-free interfacial growth of graphdiyne hollow microspheres and the mechanistic origin of their SERS activity},
author = {Lu Zhang and Wencai Yi and Junfang Li and Guoying Wei and Guangcheng Xi and Lanqun Mao},
url = {https://www.nature.com/articles/s41467-023-42038-3.pdf},
doi = {10.1038/s41467-023-42038-3},
year = {2023},
date = {2023-10-09},
urldate = {2023-10-09},
journal = {Nature Communications},
volume = {14},
number = {6318},
abstract = {As a two-dimensional carbon allotrope, graphdiyne possesses a direct band gap, excellent charge carrier mobility, and uniformly distributed pores. Here, a surfactant-free growth method is developed to efficiently synthesize graphdiyne hollow microspheres at liquid‒liquid interfaces with a self-supporting structure, which avoids the influence of surfactants on product properties. We demonstrate that pristine graphdiyne hollow microspheres, without any additional functionalization, show a strong surface-enhanced Raman scattering effect with an enhancement factor of 3.7 × 10^7 and a detection limit of 1 × 10^−12 M for rhodamine 6 G, which is approximately 1000 times that of graphene. Experimental measurements and first-principles density functional theory simulations confirm the hypothesis that the surface-enhanced Raman scattering activity can be attributed to an efficiency interfacial charge transfer within the graphdiyne-molecule system.},
note = {The 2D carbon allotrope graphdiyne possesses a direct band gap, high charge carrier mobility, and uniformly distributed pores. Here, the authors report the surfactant-free growth of graphdiyne hollow microspheres and investigate the origin of their SERS activity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
As a two-dimensional carbon allotrope, graphdiyne possesses a direct band gap, excellent charge carrier mobility, and uniformly distributed pores. Here, a surfactant-free growth method is developed to efficiently synthesize graphdiyne hollow microspheres at liquid‒liquid interfaces with a self-supporting structure, which avoids the influence of surfactants on product properties. We demonstrate that pristine graphdiyne hollow microspheres, without any additional functionalization, show a strong surface-enhanced Raman scattering effect with an enhancement factor of 3.7 × 10^7 and a detection limit of 1 × 10^−12 M for rhodamine 6 G, which is approximately 1000 times that of graphene. Experimental measurements and first-principles density functional theory simulations confirm the hypothesis that the surface-enhanced Raman scattering activity can be attributed to an efficiency interfacial charge transfer within the graphdiyne-molecule system. |
| 88. |  | Guomin Zhu, Benjamin A. Legg, Michel Sassi, Xinran Liang, Meirong Zong, Kevin M. Rosso, James J. De Yoreo Crystal dissolution by particle detachment In: Nature Communications, vol. 14, no. 6300, 2023, (Crystal dissolution has been predominately viewed as a process of ion-by-ion detachment into a surrounding solvent. Here, the authors report an alternative mechanism of dissolution by particle detachment.). @article{nokey,
title = {Crystal dissolution by particle detachment},
author = {Guomin Zhu and Benjamin A. Legg and Michel Sassi and Xinran Liang and Meirong Zong and Kevin M. Rosso and James J. De Yoreo },
url = {https://www.nature.com/articles/s41467-023-41443-y.pdf},
doi = {10.1038/s41467-023-41443-y},
year = {2023},
date = {2023-10-09},
urldate = {2023-10-09},
journal = {Nature Communications},
volume = {14},
number = {6300},
abstract = {Crystal dissolution, which is a fundamental process in both natural and technological settings, has been predominately viewed as a process of ion-by-ion detachment into a surrounding solvent. Here we report a mechanism of dissolution by particle detachment (DPD) that dominates in mesocrystals formed via crystallization by particle attachment (CPA). Using liquid phase electron microscopy to directly observe dissolution of hematite crystals — both compact rhombohedra and mesocrystals of coaligned nanoparticles — we find that the mesocrystals evolve into branched structures, which disintegrate as individual sub-particles detach. The resulting dissolution rates far exceed those for equivalent masses of compact single crystals. Applying a numerical generalization of the Gibbs-Thomson effect, we show that the physical drivers of DPD are curvature and strain inherently tied to the original CPA process. Based on the generality of the model, we anticipate that DPD is widespread for both natural minerals and synthetic crystals formed via CPA.},
note = {Crystal dissolution has been predominately viewed as a process of ion-by-ion detachment into a surrounding solvent. Here, the authors report an alternative mechanism of dissolution by particle detachment.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Crystal dissolution, which is a fundamental process in both natural and technological settings, has been predominately viewed as a process of ion-by-ion detachment into a surrounding solvent. Here we report a mechanism of dissolution by particle detachment (DPD) that dominates in mesocrystals formed via crystallization by particle attachment (CPA). Using liquid phase electron microscopy to directly observe dissolution of hematite crystals — both compact rhombohedra and mesocrystals of coaligned nanoparticles — we find that the mesocrystals evolve into branched structures, which disintegrate as individual sub-particles detach. The resulting dissolution rates far exceed those for equivalent masses of compact single crystals. Applying a numerical generalization of the Gibbs-Thomson effect, we show that the physical drivers of DPD are curvature and strain inherently tied to the original CPA process. Based on the generality of the model, we anticipate that DPD is widespread for both natural minerals and synthetic crystals formed via CPA. |
| 87. |  | Jeffrey R. Reimers, Tiexin Li, André P. Birvé, Likun Yang, Albert C. Aragonès, Thomas Fallon, Daniel S. Kosov, Nadim Darwish Controlling piezoresistance in single molecules through the isomerisation of bullvalenes In: Nature Communications, vol. 14, no. 6089 , 2023, (The quest for miniaturization of electromechanical nanosystems requires the use of single molecules as active components. Here, the authors develop a piezoresistor based on a single bullvalene molecule that changes its shape by a Cope rearrangement. ). @article{nokey,
title = {Controlling piezoresistance in single molecules through the isomerisation of bullvalenes},
author = {Jeffrey R. Reimers and Tiexin Li and André P. Birvé and Likun Yang and Albert C. Aragonès and Thomas Fallon and Daniel S. Kosov and Nadim Darwish},
url = {https://www.nature.com/articles/s41467-023-41674-z.pdf},
doi = {10.1038/s41467-023-41674-z},
year = {2023},
date = {2023-10-03},
urldate = {2023-10-03},
journal = {Nature Communications},
volume = {14},
number = {6089 },
abstract = {Nanoscale electro-mechanical systems (NEMS) displaying piezoresistance offer unique measurement opportunities at the sub-cellular level, in detectors and sensors, and in emerging generations of integrated electronic devices. Here, we show a single-molecule NEMS piezoresistor that operates utilising constitutional and conformational isomerisation of individual diaryl-bullvalene molecules and can be switched at 850 Hz. Observations are made using scanning tunnelling microscopy break junction (STMBJ) techniques to characterise piezoresistance, combined with blinking (current-time) experiments that follow single-molecule reactions in real time. A kinetic Monte Carlo methodology (KMC) is developed to simulate isomerisation on the experimental timescale, parameterised using density-functional theory (DFT) combined with non-equilibrium Green’s function (NEGF) calculations. Results indicate that piezoresistance is controlled by both constitutional and conformational isomerisation, occurring at rates that are either fast (equilibrium) or slow (non-equilibrium) compared to the experimental timescale. Two different types of STMBJ traces are observed, one typical of traditional experiments that are interpreted in terms of intramolecular isomerisation occurring on stable tipped-shaped metal-contact junctions, and another attributed to arise from junction‒interface restructuring induced by bullvalene isomerisation.},
note = {The quest for miniaturization of electromechanical nanosystems requires the use of single molecules as active components. Here, the authors develop a piezoresistor based on a single bullvalene molecule that changes its shape by a Cope rearrangement. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanoscale electro-mechanical systems (NEMS) displaying piezoresistance offer unique measurement opportunities at the sub-cellular level, in detectors and sensors, and in emerging generations of integrated electronic devices. Here, we show a single-molecule NEMS piezoresistor that operates utilising constitutional and conformational isomerisation of individual diaryl-bullvalene molecules and can be switched at 850 Hz. Observations are made using scanning tunnelling microscopy break junction (STMBJ) techniques to characterise piezoresistance, combined with blinking (current-time) experiments that follow single-molecule reactions in real time. A kinetic Monte Carlo methodology (KMC) is developed to simulate isomerisation on the experimental timescale, parameterised using density-functional theory (DFT) combined with non-equilibrium Green’s function (NEGF) calculations. Results indicate that piezoresistance is controlled by both constitutional and conformational isomerisation, occurring at rates that are either fast (equilibrium) or slow (non-equilibrium) compared to the experimental timescale. Two different types of STMBJ traces are observed, one typical of traditional experiments that are interpreted in terms of intramolecular isomerisation occurring on stable tipped-shaped metal-contact junctions, and another attributed to arise from junction‒interface restructuring induced by bullvalene isomerisation. |
| 86. |  | Zhenzhong Yang, Le Wang, Jeffrey A. Dhas, Mark H. Engelhard, Mark E. Bowden, Wen Liu, Zihua Zhu, Chongmin Wang, Scott A. Chambers, Peter V. Sushko, Yingge Du Guided anisotropic oxygen transport in vacancy ordered oxides In: Nature Communications, vol. 14, no. 6068, 2023, (Control of ion diffusion is instrumental in understanding the behavior and structural transformations of transition metal oxides. Here, the authors use in-situ TEM to reveal the atomic-scale evolution of the topotactic phase transition in BM-SFO triggered by the migration of oxygen ions under an external stimulus.). @article{nokey,
title = {Guided anisotropic oxygen transport in vacancy ordered oxides},
author = {Zhenzhong Yang and Le Wang and Jeffrey A. Dhas and Mark H. Engelhard and Mark E. Bowden and Wen Liu and Zihua Zhu and Chongmin Wang and Scott A. Chambers and Peter V. Sushko and Yingge Du },
url = {https://www.nature.com/articles/s41467-023-40746-4.pdf},
doi = {10.1038/s41467-023-40746-4},
year = {2023},
date = {2023-09-28},
urldate = {2023-09-28},
journal = {Nature Communications},
volume = {14},
number = {6068},
abstract = {Anisotropic and efficient transport of ions under external stimuli governs the operation and failure mechanisms of energy-conversion systems and microelectronics devices. However, fundamental understanding of ion hopping processes is impeded by the lack of atomically precise materials and probes that allow for the monitoring and control at the appropriate time- and length- scales. In this work, using in-situ transmission electron microscopy, we directly show that oxygen ion migration in vacancy ordered, semiconducting SrFeO2.5 epitaxial thin films can be guided to proceed through two distinctly different diffusion pathways, each resulting in different polymorphs of SrFeO2.75 with different ground electronic properties before reaching a fully oxidized, metallic SrFeO3 phase. The diffusion steps and reaction intermediates are revealed by means of ab-initio calculations. The principles of controlling oxygen diffusion pathways and reaction intermediates demonstrated here may advance the rational design of structurally ordered oxides for tailored applications and provide insights for developing devices with multiple states of regulation.},
note = {Control of ion diffusion is instrumental in understanding the behavior and structural transformations of transition metal oxides. Here, the authors use in-situ TEM to reveal the atomic-scale evolution of the topotactic phase transition in BM-SFO triggered by the migration of oxygen ions under an external stimulus.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Anisotropic and efficient transport of ions under external stimuli governs the operation and failure mechanisms of energy-conversion systems and microelectronics devices. However, fundamental understanding of ion hopping processes is impeded by the lack of atomically precise materials and probes that allow for the monitoring and control at the appropriate time- and length- scales. In this work, using in-situ transmission electron microscopy, we directly show that oxygen ion migration in vacancy ordered, semiconducting SrFeO2.5 epitaxial thin films can be guided to proceed through two distinctly different diffusion pathways, each resulting in different polymorphs of SrFeO2.75 with different ground electronic properties before reaching a fully oxidized, metallic SrFeO3 phase. The diffusion steps and reaction intermediates are revealed by means of ab-initio calculations. The principles of controlling oxygen diffusion pathways and reaction intermediates demonstrated here may advance the rational design of structurally ordered oxides for tailored applications and provide insights for developing devices with multiple states of regulation. |
| 85. |  | Chao Li, Christoph Kaspar, Ping Zhou, Jung-Ching Liu, Outhmane Chahib, Thilo Glatzel, Robert Häner, Ulrich Aschauer, Silvio Decurtins, Shi-Xia Liu, Michael Thoss, Ernst Meyer, Rémy Pawlak Strong signature of electron-vibration coupling in molecules on Ag(111) triggered by tip-gated discharging In: Nature Communications, vol. 14, no. 5956, 2023, (Electron-vibration coupling is driving advances in molecular electronics, spintronics, and quantum technology. Here, the authors succeeded in directly controlling vibronic excitations in tetrabromotetraazapyrene (TBTAP) molecules on the surface of Ag(111).). @article{nokey,
title = {Strong signature of electron-vibration coupling in molecules on Ag(111) triggered by tip-gated discharging},
author = {Chao Li and Christoph Kaspar and Ping Zhou and Jung-Ching Liu and Outhmane Chahib and Thilo Glatzel and Robert Häner and Ulrich Aschauer and Silvio Decurtins and Shi-Xia Liu and Michael Thoss and Ernst Meyer and Rémy Pawlak },
url = {https://www.nature.com/articles/s41467-023-41601-2.pdf},
doi = {10.1038/s41467-023-41601-2},
year = {2023},
date = {2023-09-25},
urldate = {2023-09-25},
journal = {Nature Communications},
volume = {14},
number = {5956},
abstract = {Electron-vibration coupling is of critical importance for the development of molecular electronics, spintronics, and quantum technologies, as it affects transport properties and spin dynamics. The control over charge-state transitions and subsequent molecular vibrations using scanning tunneling microscopy typically requires the use of a decoupling layer. Here we show the vibronic excitations of tetrabromotetraazapyrene (TBTAP) molecules directly adsorbed on Ag(111) into an orientational glassy phase. The electron-deficient TBTAP is singly-occupied by an electron donated from the substrate, resulting in a spin 1/2 state, which is confirmed by a Kondo resonance. The TBTAP•− discharge is controlled by tip-gating and leads to a series of peaks in scanning tunneling spectroscopy. These occurrences are explained by combining a double-barrier tunneling junction with a Franck-Condon model including molecular vibrational modes. This work demonstrates that suitable precursor design enables gate-dependent vibrational excitations of molecules on a metal, thereby providing a method to investigate electron-vibration coupling in molecular assemblies without a decoupling layer.},
note = {Electron-vibration coupling is driving advances in molecular electronics, spintronics, and quantum technology. Here, the authors succeeded in directly controlling vibronic excitations in tetrabromotetraazapyrene (TBTAP) molecules on the surface of Ag(111).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Electron-vibration coupling is of critical importance for the development of molecular electronics, spintronics, and quantum technologies, as it affects transport properties and spin dynamics. The control over charge-state transitions and subsequent molecular vibrations using scanning tunneling microscopy typically requires the use of a decoupling layer. Here we show the vibronic excitations of tetrabromotetraazapyrene (TBTAP) molecules directly adsorbed on Ag(111) into an orientational glassy phase. The electron-deficient TBTAP is singly-occupied by an electron donated from the substrate, resulting in a spin 1/2 state, which is confirmed by a Kondo resonance. The TBTAP•− discharge is controlled by tip-gating and leads to a series of peaks in scanning tunneling spectroscopy. These occurrences are explained by combining a double-barrier tunneling junction with a Franck-Condon model including molecular vibrational modes. This work demonstrates that suitable precursor design enables gate-dependent vibrational excitations of molecules on a metal, thereby providing a method to investigate electron-vibration coupling in molecular assemblies without a decoupling layer. |
| 84. |  | Pengcheng Chen, Qiuhao Xu, Zijing Ding, Qing Chen, Jiyu Xu, Zhihai Cheng, Xiaohui Qiu, Bingkai Yuan, Sheng Meng, Nan Yao Identification of a common ice nucleus on hydrophilic and hydrophobic close-packed metal surfaces In: Nature Communications, vol. 14, no. 5813, 2023, (One key challenge in water science is to create a universal model that can predict the arrangement of water/solid interfaces. Here, the authors use STM and noncontact-AFM to discover common water clusters during the initial growth on hydrophilic and hydrophobic close-packed metal surfaces.). @article{nokey,
title = {Identification of a common ice nucleus on hydrophilic and hydrophobic close-packed metal surfaces},
author = {Pengcheng Chen and Qiuhao Xu and Zijing Ding and Qing Chen and Jiyu Xu and Zhihai Cheng and Xiaohui Qiu and Bingkai Yuan and Sheng Meng and Nan Yao },
url = {https://www.nature.com/articles/s41467-023-41436-x.pdf},
doi = {10.1038/s41467-023-41436-x},
year = {2023},
date = {2023-09-19},
urldate = {2023-09-19},
journal = {Nature Communications},
volume = {14},
number = {5813},
abstract = {Establishing a general model of heterogeneous ice nucleation has long been challenging because of the surface water structures found on different substrates. Identifying common water clusters, regardless of the underlying substrate, is one of the key steps toward solving this problem. Here, we demonstrate the presence of a common water cluster found on both hydrophilic Pt(111) and hydrophobic Cu(111) surfaces using scanning tunneling microscopy and non-contact atomic force microscopy. Water molecules self-assemble into a structure with a central flat-lying hexagon and three fused pentagonal rings, forming a cluster consisting of 15 individual water molecules. This cluster serves as a critical nucleus during ice nucleation on both surfaces: ice growth beyond this cluster bifurcates to form two-dimensional (three-dimensional) layers on hydrophilic (hydrophobic) surfaces. Our results reveal the inherent similarity and distinction at the initial stage of ice growth on hydrophilic and hydrophobic close-packed metal surfaces; thus, these observations provide initial evidence toward a general model for water-substrate interaction.},
note = {One key challenge in water science is to create a universal model that can predict the arrangement of water/solid interfaces. Here, the authors use STM and noncontact-AFM to discover common water clusters during the initial growth on hydrophilic and hydrophobic close-packed metal surfaces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Establishing a general model of heterogeneous ice nucleation has long been challenging because of the surface water structures found on different substrates. Identifying common water clusters, regardless of the underlying substrate, is one of the key steps toward solving this problem. Here, we demonstrate the presence of a common water cluster found on both hydrophilic Pt(111) and hydrophobic Cu(111) surfaces using scanning tunneling microscopy and non-contact atomic force microscopy. Water molecules self-assemble into a structure with a central flat-lying hexagon and three fused pentagonal rings, forming a cluster consisting of 15 individual water molecules. This cluster serves as a critical nucleus during ice nucleation on both surfaces: ice growth beyond this cluster bifurcates to form two-dimensional (three-dimensional) layers on hydrophilic (hydrophobic) surfaces. Our results reveal the inherent similarity and distinction at the initial stage of ice growth on hydrophilic and hydrophobic close-packed metal surfaces; thus, these observations provide initial evidence toward a general model for water-substrate interaction. |
| 83. |  | Yingying Zou, Chao Liu, Chaoqi Zhang, Ling Yuan, Jiaxin Li, Tong Bao, Guangfeng Wei, Jin Zou, Chengzhong Yu Epitaxial growth of metal-organic framework nanosheets into single-crystalline orthogonal arrays In: Nature Communications, vol. 14, no. 5780, 2023, (Constructing regular 3D superstructures from 2D nanosheets is a challenge. Here, the authors report the epitaxial growth of MOF nanosheets into single-crystalline orthogonal arrays with enhanced functional properties.). @article{nokey,
title = {Epitaxial growth of metal-organic framework nanosheets into single-crystalline orthogonal arrays},
author = {Yingying Zou and Chao Liu and Chaoqi Zhang and Ling Yuan and Jiaxin Li and Tong Bao and Guangfeng Wei and Jin Zou and Chengzhong Yu },
url = {https://www.nature.com/articles/s41467-023-41517-x.pdf},
doi = {10.1038/s41467-023-41517-x},
year = {2023},
date = {2023-09-18},
urldate = {2023-09-18},
journal = {Nature Communications},
volume = {14},
number = {5780},
abstract = {Construction of two-dimensional nanosheets into three-dimensional regular structures facilitates the mass transfer and exploits the maximum potential of two-dimensional building blocks in applications such as catalysis. Here, we report the synthesis of metal-organic frameworks with an orthogonal nanosheet array. The assembly involves the epitaxial growth of single crystalline metal-organic framework nanosheets with a naturally non-preferred facet exposure as the shell on a cubic metal-organic framework as the core. The nanosheets, despite of two typical shapes and crystallographic orientations, also form a single crystalline orthogonally arrayed framework. The density and size of nanosheets in the core-shell-structured composite metal-organic frameworks can be well adjusted. Moreover, metal-organic frameworks with a single composition and hollow orthogonal nanosheet array morphology can be obtained. Benefiting from the unusual facet exposure and macroporous structure, the designed structure exhibits improved electrocatalytic oxygen evolution activity compared to conventional nanosheets.},
note = {Constructing regular 3D superstructures from 2D nanosheets is a challenge. Here, the authors report the epitaxial growth of MOF nanosheets into single-crystalline orthogonal arrays with enhanced functional properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Construction of two-dimensional nanosheets into three-dimensional regular structures facilitates the mass transfer and exploits the maximum potential of two-dimensional building blocks in applications such as catalysis. Here, we report the synthesis of metal-organic frameworks with an orthogonal nanosheet array. The assembly involves the epitaxial growth of single crystalline metal-organic framework nanosheets with a naturally non-preferred facet exposure as the shell on a cubic metal-organic framework as the core. The nanosheets, despite of two typical shapes and crystallographic orientations, also form a single crystalline orthogonally arrayed framework. The density and size of nanosheets in the core-shell-structured composite metal-organic frameworks can be well adjusted. Moreover, metal-organic frameworks with a single composition and hollow orthogonal nanosheet array morphology can be obtained. Benefiting from the unusual facet exposure and macroporous structure, the designed structure exhibits improved electrocatalytic oxygen evolution activity compared to conventional nanosheets. |
| 82. |  | Katharina Kaiser, Leonard-Alexander Lieske, Jascha Repp, Leo Gross
Charge-state lifetimes of single molecules on few monolayers of NaCl In: Nature Communications, vol. 14, no. 4988, 2023, (Resonant charge transport to and from molecules and their corresponding charge-state transitions are critical to understanding electrically driven processes. Here, the authors investigate the charge-state lifetimes of single molecules through NaCl films of 3 to 5 monolayers thickness.). @article{nokey,
title = {Charge-state lifetimes of single molecules on few monolayers of NaCl},
author = {Katharina Kaiser and Leonard-Alexander Lieske and Jascha Repp and Leo Gross
},
url = {https://www.nature.com/articles/s41467-023-40692-1.pdf},
doi = {10.1038/s41467-023-40692-1},
year = {2023},
date = {2023-08-17},
urldate = {2023-08-17},
journal = {Nature Communications},
volume = {14},
number = {4988},
abstract = {In molecular tunnel junctions, where the molecule is decoupled from the electrodes by few-monolayers-thin insulating layers, resonant charge transport takes place by sequential charge transfer to and from the molecule which implies transient charging of the molecule. The corresponding charge state transitions, which involve tunneling through the insulating decoupling layers, are crucial for understanding electrically driven processes such as electroluminescence or photocurrent generation in such a geometry. Here, we use scanning tunneling microscopy to investigate the decharging of single ZnPc and H2Pc molecules through NaCl films of 3 to 5 monolayers thickness on Cu(111) and Au(111). To this end, we approach the tip to the molecule at resonant tunnel conditions up to a regime where charge transport is limited by tunneling through the NaCl film. The resulting saturation of the tunnel current is a direct measure of the lifetimes of the anionic and cationic states, i.e., the molecule’s charge-state lifetime, and thus provides a means to study charge dynamics and, thereby, exciton dynamics. Comparison of anion and cation lifetimes on different substrates reveals the critical role of the level alignment with the insulator’s conduction and valence band, and the metal-insulator interface state.},
note = {Resonant charge transport to and from molecules and their corresponding charge-state transitions are critical to understanding electrically driven processes. Here, the authors investigate the charge-state lifetimes of single molecules through NaCl films of 3 to 5 monolayers thickness.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In molecular tunnel junctions, where the molecule is decoupled from the electrodes by few-monolayers-thin insulating layers, resonant charge transport takes place by sequential charge transfer to and from the molecule which implies transient charging of the molecule. The corresponding charge state transitions, which involve tunneling through the insulating decoupling layers, are crucial for understanding electrically driven processes such as electroluminescence or photocurrent generation in such a geometry. Here, we use scanning tunneling microscopy to investigate the decharging of single ZnPc and H2Pc molecules through NaCl films of 3 to 5 monolayers thickness on Cu(111) and Au(111). To this end, we approach the tip to the molecule at resonant tunnel conditions up to a regime where charge transport is limited by tunneling through the NaCl film. The resulting saturation of the tunnel current is a direct measure of the lifetimes of the anionic and cationic states, i.e., the molecule’s charge-state lifetime, and thus provides a means to study charge dynamics and, thereby, exciton dynamics. Comparison of anion and cation lifetimes on different substrates reveals the critical role of the level alignment with the insulator’s conduction and valence band, and the metal-insulator interface state. |
| 81. |  | Daniel Medina-Lopez, Thomas Liu, Silvio Osella, Hugo Levy-Falk, Nicolas Rolland, Christine Elias, Gaspard Huber, Pranav Ticku, Loïc Rondin, Bruno Jousselme, David Beljonne, Jean-Sébastien Lauret, Stephane Campidelli Interplay of structure and photophysics of individualized rod-shaped graphene quantum dots with up to 132 sp² carbon atoms In: Nature Communications, vol. 14, no. 4728, 2023, (The use and characterization of graphene quantum dots is limited by their pronounced tendency to form aggregates. Here, the authors synthesize rod-shaped motifs of nanographenes with up to 132
sp² carbon atoms that are fully individualized, which allows the precise description of their intrinsic photophysical properties.). @article{nokey,
title = {Interplay of structure and photophysics of individualized rod-shaped graphene quantum dots with up to 132 sp² carbon atoms},
author = {Daniel Medina-Lopez and Thomas Liu and Silvio Osella and Hugo Levy-Falk and Nicolas Rolland and Christine Elias and Gaspard Huber and Pranav Ticku and Loïc Rondin and Bruno Jousselme and David Beljonne and Jean-Sébastien Lauret and Stephane Campidelli},
url = {https://www.nature.com/articles/s41467-023-40376-w.pdf},
doi = {10.1038/s41467-023-40376-w},
year = {2023},
date = {2023-08-07},
urldate = {2023-08-07},
journal = {Nature Communications},
volume = {14},
number = {4728},
abstract = {Nanographene materials are promising building blocks for the growing field of low-dimensional materials for optics, electronics and biophotonics applications. In particular, bottom-up synthesized 0D graphene quantum dots show great potential as single quantum emitters. To fully exploit their exciting properties, the graphene quantum dots must be of high purity; the key parameter for efficient purification being the solubility of the starting materials. Here, we report the synthesis of a family of highly soluble and easily processable rod-shaped graphene quantum dots with fluorescence quantum yields up to 94%. This is uncommon for a red emission. The high solubility is directly related to the design of the structure, allowing for an accurate description of the photophysical properties of the graphene quantum dots both in solution and at the single molecule level. These photophysical properties were fully predicted by quantum-chemical calculations.},
note = {The use and characterization of graphene quantum dots is limited by their pronounced tendency to form aggregates. Here, the authors synthesize rod-shaped motifs of nanographenes with up to 132
sp² carbon atoms that are fully individualized, which allows the precise description of their intrinsic photophysical properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanographene materials are promising building blocks for the growing field of low-dimensional materials for optics, electronics and biophotonics applications. In particular, bottom-up synthesized 0D graphene quantum dots show great potential as single quantum emitters. To fully exploit their exciting properties, the graphene quantum dots must be of high purity; the key parameter for efficient purification being the solubility of the starting materials. Here, we report the synthesis of a family of highly soluble and easily processable rod-shaped graphene quantum dots with fluorescence quantum yields up to 94%. This is uncommon for a red emission. The high solubility is directly related to the design of the structure, allowing for an accurate description of the photophysical properties of the graphene quantum dots both in solution and at the single molecule level. These photophysical properties were fully predicted by quantum-chemical calculations. |
| 80. |  | Yunjun Cao, Joel Mieres-Perez, Julien Frederic Rowen, Elsa Sanchez-Garcia, Wolfram Sander, Karina Morgenstern Chirality control of a single carbene molecule by tip-induced van der Waals interactions In: Nature Communications, vol. 14, no. 4500, 2023, (The control of molecular chirality is of great interest in stereochemistry and biochemistry. Here, the authors show how to alter the chirality dynamics of a single molecule through tip-induced van der Waals interactions.). @article{nokey,
title = {Chirality control of a single carbene molecule by tip-induced van der Waals interactions},
author = {Yunjun Cao and Joel Mieres-Perez and Julien Frederic Rowen and Elsa Sanchez-Garcia and Wolfram Sander and Karina Morgenstern },
url = {https://www.nature.com/articles/s41467-023-39870-y.pdf},
doi = {10.1038/s41467-023-39870-y},
year = {2023},
date = {2023-07-26},
urldate = {2023-07-26},
journal = {Nature Communications},
volume = {14},
number = {4500},
abstract = {Non-covalent interactions such as van der Waals interactions and hydrogen bonds are crucial for the chiral induction and control of molecules, but it remains difficult to study them at the single-molecule level. Here, we report a carbene molecule on a copper surface as a prototype of an anchored molecule with a facile chirality change. We examine the influence of the attractive van der Waals interactions on the chirality change by regulating the tip-molecule distance, resulting in an excess of a carbene enantiomer. Our model study provides insight into the change of molecular chirality controlled by van der Waals interactions, which is fundamental for understanding the mechanisms of chiral induction and amplification.},
note = {The control of molecular chirality is of great interest in stereochemistry and biochemistry. Here, the authors show how to alter the chirality dynamics of a single molecule through tip-induced van der Waals interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Non-covalent interactions such as van der Waals interactions and hydrogen bonds are crucial for the chiral induction and control of molecules, but it remains difficult to study them at the single-molecule level. Here, we report a carbene molecule on a copper surface as a prototype of an anchored molecule with a facile chirality change. We examine the influence of the attractive van der Waals interactions on the chirality change by regulating the tip-molecule distance, resulting in an excess of a carbene enantiomer. Our model study provides insight into the change of molecular chirality controlled by van der Waals interactions, which is fundamental for understanding the mechanisms of chiral induction and amplification. |
| 79. |  | Liang Qiao, Nia Pollard, Ravithree D. Senanayake, Zhi Yang, Minjung Kim, Arzeena S. Ali, Minh Tam Hoang, Nan Yao, Yimo Han, Rigoberto Hernandez, Andre Z. Clayborne, Matthew R. Jones Atomically precise nanoclusters predominantly seed gold nanoparticle syntheses In: Nature Communications, vol. 14, no. 4408, 2023, (In seed-mediated synthesis, small particulate precursors are added to a growth solution to initiate heterogeneous nucleation, allowing access to well-defined colloidal nanostructures. Here, the authors identify that one of the most commonly studied seed solutions is populated by atomically precise gold nanoclusters with 32 atoms.). @article{nokey,
title = {Atomically precise nanoclusters predominantly seed gold nanoparticle syntheses},
author = {Liang Qiao and Nia Pollard and Ravithree D. Senanayake and Zhi Yang and Minjung Kim and Arzeena S. Ali and Minh Tam Hoang and Nan Yao and Yimo Han and Rigoberto Hernandez and Andre Z. Clayborne and Matthew R. Jones },
url = {https://www.nature.com/articles/s41467-023-40016-3.pdf},
doi = {10.1038/s41467-023-40016-3},
year = {2023},
date = {2023-07-21},
urldate = {2023-07-21},
journal = {Nature Communications},
volume = {14},
number = {4408},
abstract = {Seed-mediated synthesis strategies, in which small gold nanoparticle precursors are added to a growth solution to initiate heterogeneous nucleation, are among the most prevalent, simple, and productive methodologies for generating well-defined colloidal anisotropic nanostructures. However, the size, structure, and chemical properties of the seeds remain poorly understood, which partially explains the lack of mechanistic understanding of many particle growth reactions. Here, we identify the majority component in the seed solution as an atomically precise gold nanocluster, consisting of a 32-atom Au core with 8 halide ligands and 12 neutral ligands constituting a bound ion pair between a halide and the cationic surfactant: Au32X8[AQA+•X-]12 (X = Cl, Br; AQA = alkyl quaternary ammonium). Ligand exchange is dynamic and versatile, occurring on the order of minutes and allowing for the formation of 48 distinct Au32 clusters with AQAX (alkyl quaternary ammonium halide) ligands. Anisotropic nanoparticle syntheses seeded with solutions enriched in Au32X8[AQA+•X-]12 show narrower size distributions and fewer impurity particle shapes, indicating the importance of this cluster as a precursor to the growth of well-defined nanostructures.},
note = {In seed-mediated synthesis, small particulate precursors are added to a growth solution to initiate heterogeneous nucleation, allowing access to well-defined colloidal nanostructures. Here, the authors identify that one of the most commonly studied seed solutions is populated by atomically precise gold nanoclusters with 32 atoms.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Seed-mediated synthesis strategies, in which small gold nanoparticle precursors are added to a growth solution to initiate heterogeneous nucleation, are among the most prevalent, simple, and productive methodologies for generating well-defined colloidal anisotropic nanostructures. However, the size, structure, and chemical properties of the seeds remain poorly understood, which partially explains the lack of mechanistic understanding of many particle growth reactions. Here, we identify the majority component in the seed solution as an atomically precise gold nanocluster, consisting of a 32-atom Au core with 8 halide ligands and 12 neutral ligands constituting a bound ion pair between a halide and the cationic surfactant: Au32X8[AQA+•X-]12 (X = Cl, Br; AQA = alkyl quaternary ammonium). Ligand exchange is dynamic and versatile, occurring on the order of minutes and allowing for the formation of 48 distinct Au32 clusters with AQAX (alkyl quaternary ammonium halide) ligands. Anisotropic nanoparticle syntheses seeded with solutions enriched in Au32X8[AQA+•X-]12 show narrower size distributions and fewer impurity particle shapes, indicating the importance of this cluster as a precursor to the growth of well-defined nanostructures. |
| 78. |  | Maximilian Sacherer, Frank Hampel, Henry Dube Diaryl-hemiindigos as visible light, pH, and heat responsive four-state switches and application in photochromic transparent polymers In: Nature Communications, vol. 14, no. 4382, 2023, (Hemiindigo derivatives can act as photoswitches, but their applicability is limited by synthetic challenges. Here, the authors report the synthesis of modified photoswitches and demonstrate four-state switching, chemical fueling, and reversible inscription into transparent polymers.). @article{nokey,
title = {Diaryl-hemiindigos as visible light, pH, and heat responsive four-state switches and application in photochromic transparent polymers},
author = {Maximilian Sacherer and Frank Hampel and Henry Dube },
url = {https://www.nature.com/articles/s41467-023-39944-x.pdf},
doi = {10.1038/s41467-023-39944-x},
year = {2023},
date = {2023-07-20},
urldate = {2023-07-20},
journal = {Nature Communications},
volume = {14},
number = {4382},
abstract = {Photoswitches are indispensable tools for responsive chemical nanosystems and are used today in almost all areas of the natural sciences. Hemiindigo (HI) derivatives have recently been introduced as potent photoswitches, but their full applicability has been hampered by the limited possibilities of their functionalization and structural modification. Here we report on a short and easy to diversify synthesis yielding diaryl-HIs bearing one additional aromatic residue at the central double bond. The resulting chromophores offer an advantageous property profile combining red-light responsiveness, high thermal bistability, strong isomer accumulations in both switching directions, strong photochromism, tunable acid responsiveness, and acid gating. With this progress, a broader structural realm becomes accessible for HI photoswitches, which can now be synthetically tailored for advanced future applications, e.g., in research on molecular machines and switches, in studies of photoisomerization mechanisms, or in the generation of smart and addressable materials. To showcase the potential of these distinct light-responsive molecular tools, we demonstrate four-state switching, chemical fueling, and reversible inscription into transparent polymers using green and red light as well as acid/base stimuli, in addition to a comprehensive photochemical study of all compounds.},
note = {Hemiindigo derivatives can act as photoswitches, but their applicability is limited by synthetic challenges. Here, the authors report the synthesis of modified photoswitches and demonstrate four-state switching, chemical fueling, and reversible inscription into transparent polymers.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Photoswitches are indispensable tools for responsive chemical nanosystems and are used today in almost all areas of the natural sciences. Hemiindigo (HI) derivatives have recently been introduced as potent photoswitches, but their full applicability has been hampered by the limited possibilities of their functionalization and structural modification. Here we report on a short and easy to diversify synthesis yielding diaryl-HIs bearing one additional aromatic residue at the central double bond. The resulting chromophores offer an advantageous property profile combining red-light responsiveness, high thermal bistability, strong isomer accumulations in both switching directions, strong photochromism, tunable acid responsiveness, and acid gating. With this progress, a broader structural realm becomes accessible for HI photoswitches, which can now be synthetically tailored for advanced future applications, e.g., in research on molecular machines and switches, in studies of photoisomerization mechanisms, or in the generation of smart and addressable materials. To showcase the potential of these distinct light-responsive molecular tools, we demonstrate four-state switching, chemical fueling, and reversible inscription into transparent polymers using green and red light as well as acid/base stimuli, in addition to a comprehensive photochemical study of all compounds. |
| 77. |  | Alexander Hensley, Thomas E. Videbæk, Hunter Seyforth, William M. Jacobs, W. Benjamin Rogers Macroscopic photonic single crystals via seeded growth of DNA-coated colloids In: Nature Communications, vol. 14, no. 4237, 2023, (DNA-programmed colloidal assembly of macroscopic crystals for photonic applications remains elusive. Here, the authors use insights from studies of nucleation and seeded growth to develop a two-step method for assembling macroscopic photonic crystals.). @article{nokey,
title = {Macroscopic photonic single crystals via seeded growth of DNA-coated colloids},
author = {Alexander Hensley and Thomas E. Videbæk and Hunter Seyforth and William M. Jacobs and W. Benjamin Rogers },
url = {https://www.nature.com/articles/s41467-023-39992-3.pdf},
doi = {10.1038/s41467-023-39992-3},
year = {2023},
date = {2023-07-15},
urldate = {2023-07-15},
journal = {Nature Communications},
volume = {14},
number = {4237},
abstract = {Photonic crystals—a class of materials whose optical properties derive from their structure in addition to their composition—can be created by self-assembling particles whose sizes are comparable to the wavelengths of visible light. Proof-of-principle studies have shown that DNA can be used to guide the self-assembly of micrometer-sized colloidal particles into fully programmable crystal structures with photonic properties in the visible spectrum. However, the extremely temperature-sensitive kinetics of micrometer-sized DNA-functionalized particles has frustrated attempts to grow large, monodisperse crystals that are required for photonic metamaterial applications. Here we describe a robust two-step protocol for self-assembling single-domain crystals that contain millions of optical-scale DNA-functionalized particles: Monodisperse crystals are initially assembled in monodisperse droplets made by microfluidics, after which they are grown to macroscopic dimensions via seeded diffusion-limited growth. We demonstrate the generality of our approach by assembling different macroscopic single-domain photonic crystals with metamaterial properties, like structural coloration, that depend on the underlying crystal structure. By circumventing the fundamental kinetic traps intrinsic to crystallization of optical-scale DNA-coated colloids, we eliminate a key barrier to engineering photonic devices from DNA-programmed materials},
note = {DNA-programmed colloidal assembly of macroscopic crystals for photonic applications remains elusive. Here, the authors use insights from studies of nucleation and seeded growth to develop a two-step method for assembling macroscopic photonic crystals.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Photonic crystals—a class of materials whose optical properties derive from their structure in addition to their composition—can be created by self-assembling particles whose sizes are comparable to the wavelengths of visible light. Proof-of-principle studies have shown that DNA can be used to guide the self-assembly of micrometer-sized colloidal particles into fully programmable crystal structures with photonic properties in the visible spectrum. However, the extremely temperature-sensitive kinetics of micrometer-sized DNA-functionalized particles has frustrated attempts to grow large, monodisperse crystals that are required for photonic metamaterial applications. Here we describe a robust two-step protocol for self-assembling single-domain crystals that contain millions of optical-scale DNA-functionalized particles: Monodisperse crystals are initially assembled in monodisperse droplets made by microfluidics, after which they are grown to macroscopic dimensions via seeded diffusion-limited growth. We demonstrate the generality of our approach by assembling different macroscopic single-domain photonic crystals with metamaterial properties, like structural coloration, that depend on the underlying crystal structure. By circumventing the fundamental kinetic traps intrinsic to crystallization of optical-scale DNA-coated colloids, we eliminate a key barrier to engineering photonic devices from DNA-programmed materials |
| 76. | 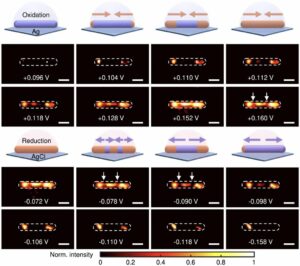 | Gang Wu, Chen Qian, Wen-Li Lv, Xiaona Zhao, Xian-Wei Liu Dynamic imaging of interfacial electrochemistry on single Ag nanowires by azimuth-modulated plasmonic scattering interferometry In: Nature Communications, vol. 14, no. 4194, 2023, (Direct visualization of the chemical dynamics of surfaces in solution is essential to gain mechanistic insights into nanocatalysis and electrochemistry. Here, the authors demonstrate the imaging of dynamic interfacial changes on a single nanowire during chemical reactions using azimuthally modulated plasmonic scattering interferometry.). @article{nokey,
title = {Dynamic imaging of interfacial electrochemistry on single Ag nanowires by azimuth-modulated plasmonic scattering interferometry},
author = {Gang Wu and Chen Qian and Wen-Li Lv and Xiaona Zhao and Xian-Wei Liu },
url = {https://www.nature.com/articles/s41467-023-39866-8.pdf},
doi = {10.1038/s41467-023-39866-8},
year = {2023},
date = {2023-07-13},
urldate = {2023-07-13},
journal = {Nature Communications},
volume = {14},
number = {4194},
abstract = {Direct visualization of surface chemical dynamics in solution is essential for understanding the mechanisms involved in nanocatalysis and electrochemistry; however, it is challenging to achieve high spatial and temporal resolution. Here, we present an azimuth-modulated plasmonic imaging technique capable of imaging dynamic interfacial changes. The method avoids strong interference from reflected light and consequently eliminates the parabolic-like interferometric patterns in the images, allowing for a 67-fold increase in the spatial resolution of plasmonic imaging. We demonstrate that this optical imaging approach enables comprehensive analyses of surface chemical dynamics and identification of previously unknown surface reaction heterogeneity by investigating electrochemical redox reactions over single silver nanowires as an example. This work provides a general strategy for high-resolution plasmonic imaging of surface electrochemical dynamics and other interfacial chemical reactions, complementing existing surface characterization methods.},
note = {Direct visualization of the chemical dynamics of surfaces in solution is essential to gain mechanistic insights into nanocatalysis and electrochemistry. Here, the authors demonstrate the imaging of dynamic interfacial changes on a single nanowire during chemical reactions using azimuthally modulated plasmonic scattering interferometry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Direct visualization of surface chemical dynamics in solution is essential for understanding the mechanisms involved in nanocatalysis and electrochemistry; however, it is challenging to achieve high spatial and temporal resolution. Here, we present an azimuth-modulated plasmonic imaging technique capable of imaging dynamic interfacial changes. The method avoids strong interference from reflected light and consequently eliminates the parabolic-like interferometric patterns in the images, allowing for a 67-fold increase in the spatial resolution of plasmonic imaging. We demonstrate that this optical imaging approach enables comprehensive analyses of surface chemical dynamics and identification of previously unknown surface reaction heterogeneity by investigating electrochemical redox reactions over single silver nanowires as an example. This work provides a general strategy for high-resolution plasmonic imaging of surface electrochemical dynamics and other interfacial chemical reactions, complementing existing surface characterization methods. |
| 75. |  | Yunqing Kang, Ovidiu Cretu, Jun Kikkawa, Koji Kimoto, Hiroki Nara, Asep Sugih Nugraha, Hiroki Kawamoto, Miharu Eguchi, Ting Liao, Ziqi Sun, Toru Asahi, Yusuke Yamauchi Mesoporous multimetallic nanospheres with exposed highly entropic alloy sites In: Nature Communications, vol. 14, no. 4182, 2023, (Nanostructured materials made from multimetal alloys are attractive for catalytic applications. Here, the authors propose a soft-templating approach to prepare mesoporous PtPdRhRuCu nanospheres that feature exposed porous structures rich in highly entropic alloy sites.). @article{nokey,
title = {Mesoporous multimetallic nanospheres with exposed highly entropic alloy sites},
author = {Yunqing Kang and Ovidiu Cretu and Jun Kikkawa and Koji Kimoto and Hiroki Nara and Asep Sugih Nugraha and Hiroki Kawamoto and Miharu Eguchi and Ting Liao and Ziqi Sun and Toru Asahi and Yusuke Yamauchi },
url = {https://www.nature.com/articles/s41467-023-39157-2.pdf},
doi = {10.1038/s41467-023-39157-2},
year = {2023},
date = {2023-07-12},
urldate = {2023-07-12},
journal = {Nature Communications},
volume = {14},
number = {4182},
abstract = {Multimetallic alloys (MMAs) with various compositions enrich the materials library with increasing diversity and have received much attention in catalysis applications. However, precisely shaping MMAs in mesoporous nanostructures and mapping the distributions of multiple elements remain big challenge due to the different reduction kinetics of various metal precursors and the complexity of crystal growth. Here we design a one-pot wet-chemical reduction approach to synthesize core–shell motif PtPdRhRuCu mesoporous nanospheres (PtPdRhRuCu MMNs) using a diblock copolymer as the soft template. The PtPdRhRuCu MMNs feature adjustable compositions and exposed porous structures rich in highly entropic alloy sites. The formation processes of the mesoporous structures and the reduction and growth kinetics of different metal precursors of PtPdRhRuCu MMNs are revealed. The PtPdRhRuCu MMNs exhibit robust electrocatalytic hydrogen evolution reaction (HER) activities and low overpotentials of 10, 13, and 28 mV at a current density of 10 mA cm−2 in alkaline (1.0 M KOH), acidic (0.5 M H2SO4), and neutral (1.0 M phosphate buffer solution (PBS)) electrolytes, respectively. The accelerated kinetics of the HER in PtPdRhRuCu MMNs are derived from multiple compositions with synergistic interactions among various metal sites and mesoporous structures with excellent mass/electron transportation characteristics.},
note = {Nanostructured materials made from multimetal alloys are attractive for catalytic applications. Here, the authors propose a soft-templating approach to prepare mesoporous PtPdRhRuCu nanospheres that feature exposed porous structures rich in highly entropic alloy sites.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Multimetallic alloys (MMAs) with various compositions enrich the materials library with increasing diversity and have received much attention in catalysis applications. However, precisely shaping MMAs in mesoporous nanostructures and mapping the distributions of multiple elements remain big challenge due to the different reduction kinetics of various metal precursors and the complexity of crystal growth. Here we design a one-pot wet-chemical reduction approach to synthesize core–shell motif PtPdRhRuCu mesoporous nanospheres (PtPdRhRuCu MMNs) using a diblock copolymer as the soft template. The PtPdRhRuCu MMNs feature adjustable compositions and exposed porous structures rich in highly entropic alloy sites. The formation processes of the mesoporous structures and the reduction and growth kinetics of different metal precursors of PtPdRhRuCu MMNs are revealed. The PtPdRhRuCu MMNs exhibit robust electrocatalytic hydrogen evolution reaction (HER) activities and low overpotentials of 10, 13, and 28 mV at a current density of 10 mA cm−2 in alkaline (1.0 M KOH), acidic (0.5 M H2SO4), and neutral (1.0 M phosphate buffer solution (PBS)) electrolytes, respectively. The accelerated kinetics of the HER in PtPdRhRuCu MMNs are derived from multiple compositions with synergistic interactions among various metal sites and mesoporous structures with excellent mass/electron transportation characteristics. |
| 74. |  | Marta Quintanilla Thermometry on individual nanoparticles highlights the impact of bimetallic interfaces In: Nature Communications, vol. 14, no. 3812, 2023, (A new study sheds light on the impact of bimetallic interfaces in nanomaterials for heat generation using single-particle thermometry. Moving from nanoparticle ensembles to single particles is key to developing consistent knowledge of material performance and nanoscale processes, but also involves assumptions and definitions that require careful consideration.). @article{nokey,
title = {Thermometry on individual nanoparticles highlights the impact of bimetallic interfaces},
author = {Marta Quintanilla},
url = {https://www.nature.com/articles/s41467-023-38983-8.pdf},
doi = {10.1038/s41467-023-38983-8},
year = {2023},
date = {2023-06-27},
urldate = {2023-06-27},
journal = {Nature Communications},
volume = {14},
number = {3812},
abstract = {A new study sheds light on the impact of bimetallic interfaces in nanomaterials for heat generation using single-particle thermometry. Moving from nanoparticle ensembles to single particles is key to developing consistent knowledge of material performance and nanoscale processes, but also involves assumptions and definitions that require careful consideration.},
note = {A new study sheds light on the impact of bimetallic interfaces in nanomaterials for heat generation using single-particle thermometry. Moving from nanoparticle ensembles to single particles is key to developing consistent knowledge of material performance and nanoscale processes, but also involves assumptions and definitions that require careful consideration.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
A new study sheds light on the impact of bimetallic interfaces in nanomaterials for heat generation using single-particle thermometry. Moving from nanoparticle ensembles to single particles is key to developing consistent knowledge of material performance and nanoscale processes, but also involves assumptions and definitions that require careful consideration. |
| 73. |  | Julian Gargiulo, Matias Herran, Ianina L. Violi, Ana Sousa-Castillo, Luciana P. Martinez, Simone Ezendam, Mariano Barella, Helene Giesler, Roland Grzeschik, Sebastian Schlücker, Stefan A. Maier, Fernando D. Stefani, Emiliano Cortés Impact of bimetallic interface design on heat generation in plasmonic Au/Pd nanostructures studied by single-particle thermometry In: Nature Communications, vol. 14, no. 3813, 2023, (The conversion of light into heat is a key process for plasmonic-catalytic applications. Here, the authors investigate how the design of the bimetallic interface affects the photothermal heating properties in Au/Pd nanostructures by applying thermometry at the single-particle level.). @article{nokey,
title = {Impact of bimetallic interface design on heat generation in plasmonic Au/Pd nanostructures studied by single-particle thermometry},
author = {Julian Gargiulo and Matias Herran and Ianina L. Violi and Ana Sousa-Castillo and Luciana P. Martinez and Simone Ezendam and Mariano Barella and Helene Giesler and Roland Grzeschik and Sebastian Schlücker and Stefan A. Maier and Fernando D. Stefani and Emiliano Cortés },
url = {https://www.nature.com/articles/s41467-023-38982-9.pdf},
doi = {10.1038/s41467-023-38982-9},
year = {2023},
date = {2023-06-26},
urldate = {2023-06-26},
journal = {Nature Communications},
volume = {14},
number = {3813},
abstract = {Localized surface plasmons are lossy and generate heat. However, accurate measurement of the temperature of metallic nanoparticles under illumination remains an open challenge, creating difficulties in the interpretation of results across plasmonic applications. Particularly, there is a quest for understanding the role of temperature in plasmon-assisted catalysis. Bimetallic nanoparticles combining plasmonic with catalytic metals are raising increasing interest in artificial photosynthesis and the production of solar fuels. Here, we perform single-particle thermometry measurements to investigate the link between morphology and light-to-heat conversion of colloidal Au/Pd nanoparticles with two different configurations: core–shell and core-satellite. It is observed that the inclusion of Pd as a shell strongly reduces the photothermal response in comparison to the bare cores, while the inclusion of Pd as satellites keeps photothermal properties almost unaffected. These results contribute to a better understanding of energy conversion processes in plasmon-assisted catalysis.},
note = {The conversion of light into heat is a key process for plasmonic-catalytic applications. Here, the authors investigate how the design of the bimetallic interface affects the photothermal heating properties in Au/Pd nanostructures by applying thermometry at the single-particle level.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Localized surface plasmons are lossy and generate heat. However, accurate measurement of the temperature of metallic nanoparticles under illumination remains an open challenge, creating difficulties in the interpretation of results across plasmonic applications. Particularly, there is a quest for understanding the role of temperature in plasmon-assisted catalysis. Bimetallic nanoparticles combining plasmonic with catalytic metals are raising increasing interest in artificial photosynthesis and the production of solar fuels. Here, we perform single-particle thermometry measurements to investigate the link between morphology and light-to-heat conversion of colloidal Au/Pd nanoparticles with two different configurations: core–shell and core-satellite. It is observed that the inclusion of Pd as a shell strongly reduces the photothermal response in comparison to the bare cores, while the inclusion of Pd as satellites keeps photothermal properties almost unaffected. These results contribute to a better understanding of energy conversion processes in plasmon-assisted catalysis. |
| 72. |  | Jiapeng Zheng, Christina Boukouvala, George R. Lewis, Yicong Ma, Yang Chen, Emilie Ringe, Lei Shao, Zhifeng Huang, Jianfang Wang Halide-assisted differential growth of chiral nanoparticles with threefold rotational symmetry In: Nature Communications, vol. 14, no. 3783, 2023, (Expanding the library of chiral plasmonic nanoparticles will foster the development of chiroptical applications. Here, the authors apply halide-assisted differential growth to convert Au nanodisks into triskelion-shaped chiral nanoparticles with threefold rotational symmetry.). @article{nokey,
title = {Halide-assisted differential growth of chiral nanoparticles with threefold rotational symmetry},
author = {Jiapeng Zheng and Christina Boukouvala and George R. Lewis and Yicong Ma and Yang Chen and Emilie Ringe and Lei Shao and Zhifeng Huang and Jianfang Wang },
url = {https://www.nature.com/articles/s41467-023-39456-8.pdf},
doi = {10.1038/s41467-023-39456-8},
year = {2023},
date = {2023-06-25},
urldate = {2023-06-25},
journal = {Nature Communications},
volume = {14},
number = {3783},
abstract = {Enriching the library of chiral plasmonic nanoparticles that can be chemically mass-produced will greatly facilitate the applications of chiral plasmonics in areas ranging from constructing optical metamaterials to sensing chiral molecules and activating immune cells. Here we report on a halide-assisted differential growth strategy that can direct the anisotropic growth of chiral Au nanoparticles with tunable sizes and diverse morphologies. Anisotropic Au nanodisks are employed as seeds to yield triskelion-shaped chiral nanoparticles with threefold rotational symmetry and high dissymmetry factors. The averaged scattering g-factors of the L- and D-nanotriskelions are as large as 0.57 and − 0.49 at 650 nm, respectively. The Au nanotriskelions have been applied in chiral optical switching devices and chiral nanoemitters. We also demonstrate that the manipulation of the directional growth rate enables the generation of a variety of chiral morphologies in the presence of homochiral ligands.
},
note = {Expanding the library of chiral plasmonic nanoparticles will foster the development of chiroptical applications. Here, the authors apply halide-assisted differential growth to convert Au nanodisks into triskelion-shaped chiral nanoparticles with threefold rotational symmetry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Enriching the library of chiral plasmonic nanoparticles that can be chemically mass-produced will greatly facilitate the applications of chiral plasmonics in areas ranging from constructing optical metamaterials to sensing chiral molecules and activating immune cells. Here we report on a halide-assisted differential growth strategy that can direct the anisotropic growth of chiral Au nanoparticles with tunable sizes and diverse morphologies. Anisotropic Au nanodisks are employed as seeds to yield triskelion-shaped chiral nanoparticles with threefold rotational symmetry and high dissymmetry factors. The averaged scattering g-factors of the L- and D-nanotriskelions are as large as 0.57 and − 0.49 at 650 nm, respectively. The Au nanotriskelions have been applied in chiral optical switching devices and chiral nanoemitters. We also demonstrate that the manipulation of the directional growth rate enables the generation of a variety of chiral morphologies in the presence of homochiral ligands.
|
| 71. |  | Kunmo Koo, Bo Shen, Sung-Il Baik, Zugang Mao, Paul J. M. Smeets, Ivan Cheuk, Kun He, Roberto dos Reis, Liliang Huang, Zihao Ye, Xiaobing Hu, Chad A. Mirkin, Vinayak P. Dravid Formation mechanism of high-index faceted Pt-Bi alloy nanoparticles by evaporation-induced growth from metal salts In: Nature Communications, vol. 14, no. 3790, 2023, (Faceted nanoparticles with high-index planes exposed usually bear promising material functionalities. Here, the authors directly observe the coarsening and facet regulation process of tetrahexahedra Pt nanoparticles by using in-situ gas-phase TEM.). @article{nokey,
title = {Formation mechanism of high-index faceted Pt-Bi alloy nanoparticles by evaporation-induced growth from metal salts},
author = {Kunmo Koo and Bo Shen and Sung-Il Baik and Zugang Mao and Paul J. M. Smeets and Ivan Cheuk and Kun He and Roberto dos Reis and Liliang Huang and Zihao Ye and Xiaobing Hu and Chad A. Mirkin and Vinayak P. Dravid },
url = {https://www.nature.com/articles/s41467-023-39458-6.pdf},
doi = {10.1038/s41467-023-39458-6},
year = {2023},
date = {2023-06-24},
urldate = {2023-06-24},
journal = {Nature Communications},
volume = {14},
number = {3790},
abstract = {Nanoparticles with high-index facets are intriguing because such facets can lend the structure useful functionality, including enhanced catalytic performance and wide-ranging optical tunability. Ligand-free solid-state syntheses of high index-facet nanoparticles, through an alloying-dealloying process with foreign volatile metals, are attractive owing to their materials generality and high yields. However, the role of foreign atoms in stabilizing the high-index facets and the dynamic nature of the transformation including the coarsening and facet regulation process are still poorly understood. Herein, the transformation of Pt salts to spherical seeds and then to tetrahexahedra, is studied in situ via gas-cell transmission electron microscopy. The dynamic behaviors of the alloying and dealloying process, which involves the coarsening of nanoparticles and consequent facet regulation stage are captured in the real time with a nanoscale spatial resolution. Based on additional direct evidence obtained using atom probe tomography and density functional theory calculations, the underlying mechanisms of the alloying-dealloying process are uncovered, which will facilitate broader explorations of high-index facet nanoparticle synthesis.},
note = {Faceted nanoparticles with high-index planes exposed usually bear promising material functionalities. Here, the authors directly observe the coarsening and facet regulation process of tetrahexahedra Pt nanoparticles by using in-situ gas-phase TEM.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanoparticles with high-index facets are intriguing because such facets can lend the structure useful functionality, including enhanced catalytic performance and wide-ranging optical tunability. Ligand-free solid-state syntheses of high index-facet nanoparticles, through an alloying-dealloying process with foreign volatile metals, are attractive owing to their materials generality and high yields. However, the role of foreign atoms in stabilizing the high-index facets and the dynamic nature of the transformation including the coarsening and facet regulation process are still poorly understood. Herein, the transformation of Pt salts to spherical seeds and then to tetrahexahedra, is studied in situ via gas-cell transmission electron microscopy. The dynamic behaviors of the alloying and dealloying process, which involves the coarsening of nanoparticles and consequent facet regulation stage are captured in the real time with a nanoscale spatial resolution. Based on additional direct evidence obtained using atom probe tomography and density functional theory calculations, the underlying mechanisms of the alloying-dealloying process are uncovered, which will facilitate broader explorations of high-index facet nanoparticle synthesis. |
| 70. |  | Hak June Lee, Seongbin Im, Dongju Jung, Kyuri Kim, Jong Ah Chae, Jaemin Lim, Jeong Woo Park, Doyoon Shin, Kookheon Char, Byeong Guk Jeong, Ji-Sang Park, Euyheon Hwang, Doh C. Lee, Young-Shin Park, Hyung-Jun Song, Jun Hyuk Chang, Wan Ki Bae Coherent heteroepitaxial growth of I-III-VI2 Ag(In,Ga)S2 colloidal nanocrystals with near-unity quantum yield for use in luminescent solar concentrators In: Nature Communications, vol. 14, no. 3779, 2023, (Colloidal semiconductor core-shell nanocrystals are sought after for photonic applications. Here, the authors report coherent heteroepitaxial growth of Ag(In,Ga)S2 core-shell nanocrystals with near-unity photoluminescence quantum yield across almost the full visible range.). @article{nokey,
title = {Coherent heteroepitaxial growth of I-III-VI2 Ag(In,Ga)S2 colloidal nanocrystals with near-unity quantum yield for use in luminescent solar concentrators},
author = {Hak June Lee and Seongbin Im and Dongju Jung and Kyuri Kim and Jong Ah Chae and Jaemin Lim and Jeong Woo Park and Doyoon Shin and Kookheon Char and Byeong Guk Jeong and Ji-Sang Park and Euyheon Hwang and Doh C. Lee and Young-Shin Park and Hyung-Jun Song and Jun Hyuk Chang and Wan Ki Bae },
url = {https://www.nature.com/articles/s41467-023-39509-y.pdf},
doi = {10.1038/s41467-023-39509-y},
year = {2023},
date = {2023-06-23},
urldate = {2023-06-23},
journal = {Nature Communications},
volume = {14},
number = {3779},
abstract = {Colloidal Ag(In,Ga)S2 nanocrystals (AIGS NCs) with the band gap tunability by their size and composition within visible range have garnered surging interest. High absorption cross-section and narrow emission linewidth of AIGS NCs make them ideally suited to address the challenges of Cd-free NCs in wide-ranging photonic applications. However, AIGS NCs have shown relatively underwhelming photoluminescence quantum yield (PL QY) to date, primarily because coherent heteroepitaxy has not been realized. Here, we report the heteroepitaxy for AIGS-AgGaS2 (AIGS-AGS) core-shell NCs bearing near-unity PL QYs in almost full visible range (460 to 620 nm) and enhanced photochemical stability. Key to the successful growth of AIGS-AGS NCs is the use of the Ag-S-Ga(OA)2 complex, which complements the reactivities among cations for both homogeneous AIGS cores in various compositions and uniform AGS shell growth. The heteroepitaxy between AIGS and AGS results in the Type I heterojunction that effectively confines charge carriers within the emissive core without optically active interfacial defects. AIGS-AGS NCs show higher extinction coefficient and narrower spectral linewidth compared to state-of-the-art heavy metal-free NCs, prompting their immediate use in practicable applications including displays and luminescent solar concentrators (LSCs).},
note = {Colloidal semiconductor core-shell nanocrystals are sought after for photonic applications. Here, the authors report coherent heteroepitaxial growth of Ag(In,Ga)S2 core-shell nanocrystals with near-unity photoluminescence quantum yield across almost the full visible range.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Colloidal Ag(In,Ga)S2 nanocrystals (AIGS NCs) with the band gap tunability by their size and composition within visible range have garnered surging interest. High absorption cross-section and narrow emission linewidth of AIGS NCs make them ideally suited to address the challenges of Cd-free NCs in wide-ranging photonic applications. However, AIGS NCs have shown relatively underwhelming photoluminescence quantum yield (PL QY) to date, primarily because coherent heteroepitaxy has not been realized. Here, we report the heteroepitaxy for AIGS-AgGaS2 (AIGS-AGS) core-shell NCs bearing near-unity PL QYs in almost full visible range (460 to 620 nm) and enhanced photochemical stability. Key to the successful growth of AIGS-AGS NCs is the use of the Ag-S-Ga(OA)2 complex, which complements the reactivities among cations for both homogeneous AIGS cores in various compositions and uniform AGS shell growth. The heteroepitaxy between AIGS and AGS results in the Type I heterojunction that effectively confines charge carriers within the emissive core without optically active interfacial defects. AIGS-AGS NCs show higher extinction coefficient and narrower spectral linewidth compared to state-of-the-art heavy metal-free NCs, prompting their immediate use in practicable applications including displays and luminescent solar concentrators (LSCs). |
| 69. |  | Chang Liu, Yan Zhao, Tai-Song Zhang, Cheng-Bo Tao, Wenwen Fei, Sheng Zhang, Man-Bo Li Asymmetric transformation of achiral gold nanoclusters with negative nonlinear dependence between chiroptical activity and enantiomeric excess In: Nature Communications, vol. 14, no. 3730, 2023, (Atomically precise metal nanoclusters provide an ideal platform for studying chirality at the nanoscale. Here, the authors use a phosphoramidite ligand as a chiral inducer to asymmetrically transform achiral into chiral gold nanoclusters with negative nonlinear CD-ee dependence.). @article{nokey,
title = {Asymmetric transformation of achiral gold nanoclusters with negative nonlinear dependence between chiroptical activity and enantiomeric excess},
author = {Chang Liu and Yan Zhao and Tai-Song Zhang and Cheng-Bo Tao and Wenwen Fei and Sheng Zhang and Man-Bo Li },
url = {https://www.nature.com/articles/s41467-023-39462-w.pdf},
doi = {10.1038/s41467-023-39462-w},
year = {2023},
date = {2023-06-22},
urldate = {2023-06-22},
journal = {Nature Communications},
volume = {14},
number = {3730},
abstract = {The investigation of chirality at the nanoscale is important to bridge the gap between molecular and macroscopic chirality. Atomically precise metal nanoclusters provide an ideal platform for this research, while their enantiopure preparation poses a challenge. Here, we describe an efficient approach to enantiopure metal nanoclusters via asymmetric transformation, that is, achiral Au23(SC6H11)16 nanoclusters are converted into chiral and enantiopure Au24(L)2(SC6H11)16 nanoclusters by a chiral inducer phosphoramidite (L). Two enantiomers of Au24(L)2(SC6H11)16 are obtained and the crystal structures reveal their hierarchical chirality, which originates from the two introduced chiral L molecules, the transformation-triggered asymmetric rearrangement of the staple motifs on the surface of the gold core, and the helical arrangement of nanocluster molecules. The construction of this type of enantiomerically pure nanoclusters is achieved based on the easy-to-synthesize and modular L. Lastly, the chirality-related chiroptical performance was investigated, revealing a negative nonlinear CD-ee dependence.},
note = {Atomically precise metal nanoclusters provide an ideal platform for studying chirality at the nanoscale. Here, the authors use a phosphoramidite ligand as a chiral inducer to asymmetrically transform achiral into chiral gold nanoclusters with negative nonlinear CD-ee dependence.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The investigation of chirality at the nanoscale is important to bridge the gap between molecular and macroscopic chirality. Atomically precise metal nanoclusters provide an ideal platform for this research, while their enantiopure preparation poses a challenge. Here, we describe an efficient approach to enantiopure metal nanoclusters via asymmetric transformation, that is, achiral Au23(SC6H11)16 nanoclusters are converted into chiral and enantiopure Au24(L)2(SC6H11)16 nanoclusters by a chiral inducer phosphoramidite (L). Two enantiomers of Au24(L)2(SC6H11)16 are obtained and the crystal structures reveal their hierarchical chirality, which originates from the two introduced chiral L molecules, the transformation-triggered asymmetric rearrangement of the staple motifs on the surface of the gold core, and the helical arrangement of nanocluster molecules. The construction of this type of enantiomerically pure nanoclusters is achieved based on the easy-to-synthesize and modular L. Lastly, the chirality-related chiroptical performance was investigated, revealing a negative nonlinear CD-ee dependence. |
| 68. |  | Nathaniel H. Park, Matteo Manica, Jannis Born, James L. Hedrick, Tim Erdmann, Dmitry Yu. Zubarev, Nil Adell-Mill, Pedro L. Arrechea Artificial intelligence driven design of catalysts and materials for ring opening polymerization using a domain-specific language In: Nature Communications, vol. 14, no. 3686, 2023, (Developing accessible polymeric materials is a challenging task for generative AI models. Here, the authors show that a domain-specific language enables the use of historical data to tune generative models to produce experimentally achievable material designs.). @article{nokey,
title = {Artificial intelligence driven design of catalysts and materials for ring opening polymerization using a domain-specific language},
author = {Nathaniel H. Park and Matteo Manica and Jannis Born and James L. Hedrick and Tim Erdmann and Dmitry Yu. Zubarev and Nil Adell-Mill and Pedro L. Arrechea },
url = {https://www.nature.com/articles/s41467-023-39396-3.pdf},
doi = {10.1038/s41467-023-39396-3},
year = {2023},
date = {2023-06-21},
urldate = {2023-06-21},
journal = {Nature Communications},
volume = {14},
number = {3686},
abstract = {Advances in machine learning (ML) and automated experimentation are poised to vastly accelerate research in polymer science. Data representation is a critical aspect for enabling ML integration in research workflows, yet many data models impose significant rigidity making it difficult to accommodate a broad array of experiment and data types found in polymer science. This inflexibility presents a significant barrier for researchers to leverage their historical data in ML development. Here we show that a domain specific language, termed Chemical Markdown Language (CMDL), provides flexible, extensible, and consistent representation of disparate experiment types and polymer structures. CMDL enables seamless use of historical experimental data to fine-tune regression transformer (RT) models for generative molecular design tasks. We demonstrate the utility of this approach through the generation and the experimental validation of catalysts and polymers in the context of ring-opening polymerization—although we provide examples of how CMDL can be more broadly applied to other polymer classes. Critically, we show how the CMDL tuned model preserves key functional groups within the polymer structure, allowing for experimental validation. These results reveal the versatility of CMDL and how it facilitates translation of historical data into meaningful predictive and generative models to produce experimentally actionable output.},
note = {Developing accessible polymeric materials is a challenging task for generative AI models. Here, the authors show that a domain-specific language enables the use of historical data to tune generative models to produce experimentally achievable material designs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Advances in machine learning (ML) and automated experimentation are poised to vastly accelerate research in polymer science. Data representation is a critical aspect for enabling ML integration in research workflows, yet many data models impose significant rigidity making it difficult to accommodate a broad array of experiment and data types found in polymer science. This inflexibility presents a significant barrier for researchers to leverage their historical data in ML development. Here we show that a domain specific language, termed Chemical Markdown Language (CMDL), provides flexible, extensible, and consistent representation of disparate experiment types and polymer structures. CMDL enables seamless use of historical experimental data to fine-tune regression transformer (RT) models for generative molecular design tasks. We demonstrate the utility of this approach through the generation and the experimental validation of catalysts and polymers in the context of ring-opening polymerization—although we provide examples of how CMDL can be more broadly applied to other polymer classes. Critically, we show how the CMDL tuned model preserves key functional groups within the polymer structure, allowing for experimental validation. These results reveal the versatility of CMDL and how it facilitates translation of historical data into meaningful predictive and generative models to produce experimentally actionable output. |
| 67. |  | Chun Tang, Thijs Stuyver, Taige Lu, Junyang Liu, Yiling Ye, Tengyang Gao, Luchun Lin, Jueting Zheng, Wenqing Liu, Jia Shi, Sason Shaik, Haiping Xia, Wenjing Hong Voltage-driven control of single-molecule keto-enol equilibrium in a two-terminal junction system In: Nature Communications, vol. 14, no. 3657, 2023, (Keto-enol tautomerism offers a promising platform for modulating charge transport at the nanoscale. Here, the authors show that the keto-enol equilibrium can be modulated on the single-molecule scale by controlling charge injection in a two-terminal junction system.). @article{nokey,
title = {Voltage-driven control of single-molecule keto-enol equilibrium in a two-terminal junction system},
author = {Chun Tang and Thijs Stuyver and Taige Lu and Junyang Liu and Yiling Ye and Tengyang Gao and Luchun Lin and Jueting Zheng and Wenqing Liu and Jia Shi and Sason Shaik and Haiping Xia and Wenjing Hong },
url = {https://www.nature.com/articles/s41467-023-39198-7.pdf},
doi = {10.1038/s41467-023-39198-7},
year = {2023},
date = {2023-06-20},
urldate = {2023-06-20},
journal = {Nature Communications},
volume = {14},
number = {3657},
abstract = {Keto-enol tautomerism, describing an equilibrium involving two tautomers with distinctive structures, provides a promising platform for modulating nanoscale charge transport. However, such equilibria are generally dominated by the keto form, while a high isomerization barrier limits the transformation to the enol form, suggesting a considerable challenge to control the tautomerism. Here, we achieve single-molecule control of a keto-enol equilibrium at room temperature by using a strategy that combines redox control and electric field modulation. Based on the control of charge injection in the single-molecule junction, we could access charged potential energy surfaces with opposite thermodynamic driving forces, i.e., exhibiting a preference for the conducting enol form, while the isomerization barrier is also significantly reduced. Thus, we could selectively obtain desired and stable tautomers, which leads to significant modulation of the single-molecule conductance. This work highlights the concept of single-molecule control of chemical reactions on more than one potential energy surface.},
note = {Keto-enol tautomerism offers a promising platform for modulating charge transport at the nanoscale. Here, the authors show that the keto-enol equilibrium can be modulated on the single-molecule scale by controlling charge injection in a two-terminal junction system.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Keto-enol tautomerism, describing an equilibrium involving two tautomers with distinctive structures, provides a promising platform for modulating nanoscale charge transport. However, such equilibria are generally dominated by the keto form, while a high isomerization barrier limits the transformation to the enol form, suggesting a considerable challenge to control the tautomerism. Here, we achieve single-molecule control of a keto-enol equilibrium at room temperature by using a strategy that combines redox control and electric field modulation. Based on the control of charge injection in the single-molecule junction, we could access charged potential energy surfaces with opposite thermodynamic driving forces, i.e., exhibiting a preference for the conducting enol form, while the isomerization barrier is also significantly reduced. Thus, we could selectively obtain desired and stable tautomers, which leads to significant modulation of the single-molecule conductance. This work highlights the concept of single-molecule control of chemical reactions on more than one potential energy surface. |
| 66. |  | Xuesong Yang, Linfeng Lan, Liang Li, Jinyang Yu, Xiaokong Liu, Ying Tao, Quan-Hong Yang, Panče Naumov, Hongyu Zhang Collective photothermal bending of flexible organic crystals modified with MXene-polymer multilayers as optical waveguide arrays In: Nature Communications, vol. 14, no. 3627, 2023, (Organic crystals represent potential alternatives to silicon dioxide due to their high flexibility and controllable properties. Here, the authors prepared a hybrid organic crystal material that responds mechanically to infrared light.). @article{nokey,
title = {Collective photothermal bending of flexible organic crystals modified with MXene-polymer multilayers as optical waveguide arrays},
author = {Xuesong Yang and Linfeng Lan and Liang Li and Jinyang Yu and Xiaokong Liu and Ying Tao and Quan-Hong Yang and Panče Naumov and Hongyu Zhang },
url = {https://www.nature.com/articles/s41467-023-39162-5.pdf},
doi = {10.1038/s41467-023-39162-5},
year = {2023},
date = {2023-06-19},
urldate = {2023-06-19},
journal = {Nature Communications},
volume = {14},
number = {3627},
abstract = {The performance of any engineering material is naturally limited by its structure, and while each material suffers from one or multiple shortcomings when considered for a particular application, these can be potentially circumvented by hybridization with other materials. By combining organic crystals with MXenes as thermal absorbers and charged polymers as adhesive counter-ionic components, we propose a simple access to flexible hybrid organic crystal materials that have the ability to mechanically respond to infrared light. The ensuing hybrid organic crystals are durable, respond fast, and can be cycled between straight and deformed state repeatedly without fatigue. The point of flexure and the curvature of the crystals can be precisely controlled by modulating the position, duration, and power of thermal excitation, and this control can be extended from individual hybrid crystals to motion of ordered two-dimensional arrays of such crystals. We also demonstrate that excitation can be achieved over very long distances (>3 m). The ability to control the shape with infrared light adds to the versatility in the anticipated applications of organic crystals, most immediately in their application as thermally controllable flexible optical waveguides for signal transmission in flexible organic electronics.},
note = {Organic crystals represent potential alternatives to silicon dioxide due to their high flexibility and controllable properties. Here, the authors prepared a hybrid organic crystal material that responds mechanically to infrared light.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The performance of any engineering material is naturally limited by its structure, and while each material suffers from one or multiple shortcomings when considered for a particular application, these can be potentially circumvented by hybridization with other materials. By combining organic crystals with MXenes as thermal absorbers and charged polymers as adhesive counter-ionic components, we propose a simple access to flexible hybrid organic crystal materials that have the ability to mechanically respond to infrared light. The ensuing hybrid organic crystals are durable, respond fast, and can be cycled between straight and deformed state repeatedly without fatigue. The point of flexure and the curvature of the crystals can be precisely controlled by modulating the position, duration, and power of thermal excitation, and this control can be extended from individual hybrid crystals to motion of ordered two-dimensional arrays of such crystals. We also demonstrate that excitation can be achieved over very long distances (>3 m). The ability to control the shape with infrared light adds to the versatility in the anticipated applications of organic crystals, most immediately in their application as thermally controllable flexible optical waveguides for signal transmission in flexible organic electronics. |
| 65. | 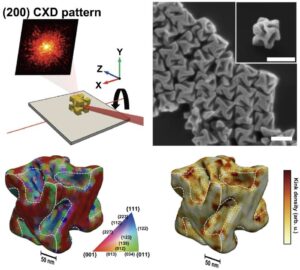 | Sungwook Choi, Sang Won Im, Ji-Hyeok Huh, Sungwon Kim, Jaeseung Kim, Yae-Chan Lim, Ryeong Myeong Kim, Jeong Hyun Han, Hyeohn Kim, Michael Sprung, Su Yong Lee, Wonsuk Cha, Ross Harder, Seungwoo Lee, Ki Tae Nam, Hyunjung Kim Strain and crystallographic identification of the helically concaved gap surfaces of chiral nanoparticles In: Nature Communications, vol. 14, no. 3615 , 2023, (Identifying the 3D crystal plane and strain field distributions of nanocrystals with concave surfaces is challenging. Here, the authors analyze the hidden concave gaps of chiral nanoparticles by using coherent Bragg X-ray diffraction imaging.). @article{nokey,
title = {Strain and crystallographic identification of the helically concaved gap surfaces of chiral nanoparticles},
author = {Sungwook Choi and Sang Won Im and Ji-Hyeok Huh and Sungwon Kim and Jaeseung Kim and Yae-Chan Lim and Ryeong Myeong Kim and Jeong Hyun Han and Hyeohn Kim and Michael Sprung and Su Yong Lee and Wonsuk Cha and Ross Harder and Seungwoo Lee and Ki Tae Nam and Hyunjung Kim },
url = {https://www.nature.com/articles/s41467-023-39255-1.pdf},
doi = {10.1038/s41467-023-39255-1},
year = {2023},
date = {2023-06-17},
urldate = {2023-06-17},
journal = {Nature Communications},
volume = {14},
number = {3615 },
abstract = {Identifying the three-dimensional (3D) crystal plane and strain-field distributions of nanocrystals is essential for optical, catalytic, and electronic applications. However, it remains a challenge to image concave surfaces of nanoparticles. Here, we develop a methodology for visualizing the 3D information of chiral gold nanoparticles ≈ 200 nm in size with concave gap structures by Bragg coherent X-ray diffraction imaging. The distribution of the high-Miller-index planes constituting the concave chiral gap is precisely determined. The highly strained region adjacent to the chiral gaps is resolved, which was correlated to the 432-symmetric morphology of the nanoparticles and its corresponding plasmonic properties are numerically predicted from the atomically defined structures. This approach can serve as a comprehensive characterization platform for visualizing the 3D crystallographic and strain distributions of nanoparticles with a few hundred nanometers, especially for applications where structural complexity and local heterogeneity are major determinants, as exemplified in plasmonics.},
note = {Identifying the 3D crystal plane and strain field distributions of nanocrystals with concave surfaces is challenging. Here, the authors analyze the hidden concave gaps of chiral nanoparticles by using coherent Bragg X-ray diffraction imaging.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Identifying the three-dimensional (3D) crystal plane and strain-field distributions of nanocrystals is essential for optical, catalytic, and electronic applications. However, it remains a challenge to image concave surfaces of nanoparticles. Here, we develop a methodology for visualizing the 3D information of chiral gold nanoparticles ≈ 200 nm in size with concave gap structures by Bragg coherent X-ray diffraction imaging. The distribution of the high-Miller-index planes constituting the concave chiral gap is precisely determined. The highly strained region adjacent to the chiral gaps is resolved, which was correlated to the 432-symmetric morphology of the nanoparticles and its corresponding plasmonic properties are numerically predicted from the atomically defined structures. This approach can serve as a comprehensive characterization platform for visualizing the 3D crystallographic and strain distributions of nanoparticles with a few hundred nanometers, especially for applications where structural complexity and local heterogeneity are major determinants, as exemplified in plasmonics. |
| 64. | 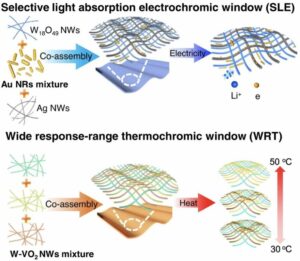 | Si-Zhe Sheng, Jin-Long Wang, Bin Zhao, Zhen He, Xue-Fei Feng, Qi-Guo Shang, Cheng Chen, Gang Pei, Jun Zhou, Jian-Wei Liu, Shu-Hong Yu Nanowire-based smart windows combining electro- and thermochromics for dynamic regulation of solar radiation In: Nature Communications, vol. 14, no. 3231, 2023, (Smart windows offer more efficient sunlight modulation and heat management. Here, the authors propose a co-assembly strategy to produce smart windows that combine electrochromic and thermochromic functions with tunable components and ordered structures for dynamic solar radiation regulation.). @article{nokey,
title = {Nanowire-based smart windows combining electro- and thermochromics for dynamic regulation of solar radiation},
author = {Si-Zhe Sheng and Jin-Long Wang and Bin Zhao and Zhen He and Xue-Fei Feng and Qi-Guo Shang and Cheng Chen and Gang Pei and Jun Zhou and Jian-Wei Liu and Shu-Hong Yu },
url = {https://www.nature.com/articles/s41467-023-38353-4.pdf},
doi = {10.1038/s41467-023-38353-4},
year = {2023},
date = {2023-06-03},
urldate = {2023-06-03},
journal = {Nature Communications},
volume = {14},
number = {3231},
abstract = {Smart window is an attractive option for efficient heat management to minimize energy consumption and improve indoor living comfort owing to their optical properties of adjusting sunlight. To effectively improve the sunlight modulation and heat management capability of smart windows, here, we propose a co-assembly strategy to fabricate the electrochromic and thermochromic smart windows with tunable components and ordered structures for the dynamic regulation of solar radiation. Firstly, to enhance both illumination and cooling efficiency in electrochromic windows, the aspect ratio and mixed type of Au nanorods are tuned to selectively absorb the near-infrared wavelength range of 760 to 1360 nm. Furthermore, when assembled with electrochromic W18O49 nanowires in the colored state, the Au nanorods exhibit a synergistic effect, resulting in a 90% reduction of near-infrared light and a corresponding 5 °C cooling effect under 1-sun irradiation. Secondly, to extend the fixed response temperature value to a wider range of 30–50 °C in thermochromic windows, the doping amount and mixed type of W-VO2 nanowires are carefully regulated. Last but not the least, the ordered assembly structure of the nanowires can greatly reduce the level of haze and enhance visibility in the windows.},
note = {Smart windows offer more efficient sunlight modulation and heat management. Here, the authors propose a co-assembly strategy to produce smart windows that combine electrochromic and thermochromic functions with tunable components and ordered structures for dynamic solar radiation regulation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Smart window is an attractive option for efficient heat management to minimize energy consumption and improve indoor living comfort owing to their optical properties of adjusting sunlight. To effectively improve the sunlight modulation and heat management capability of smart windows, here, we propose a co-assembly strategy to fabricate the electrochromic and thermochromic smart windows with tunable components and ordered structures for the dynamic regulation of solar radiation. Firstly, to enhance both illumination and cooling efficiency in electrochromic windows, the aspect ratio and mixed type of Au nanorods are tuned to selectively absorb the near-infrared wavelength range of 760 to 1360 nm. Furthermore, when assembled with electrochromic W18O49 nanowires in the colored state, the Au nanorods exhibit a synergistic effect, resulting in a 90% reduction of near-infrared light and a corresponding 5 °C cooling effect under 1-sun irradiation. Secondly, to extend the fixed response temperature value to a wider range of 30–50 °C in thermochromic windows, the doping amount and mixed type of W-VO2 nanowires are carefully regulated. Last but not the least, the ordered assembly structure of the nanowires can greatly reduce the level of haze and enhance visibility in the windows. |
| 63. | 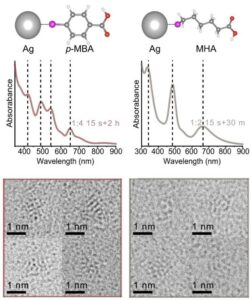 | Ji Soo Kim, Hogeun Chang, Sungsu Kang, Seungwoo Cha, Hanguk Cho, Seung Jae Kwak, Namjun Park, Younhwa Kim, Dohun Kang, Chyan Kyung Song, Jimin Kwag, Ji-Sook Hahn, Won Bo Lee, Taeghwan Hyeon, Jungwon Park Critical roles of metal–ligand complexes in the controlled synthesis of various metal nanoclusters In: Nature Communications, vol. 14, no. 3201, 2023, (Metal-thiolate complexes are the precursors to atomically precise metal nanoclusters. Here, the authors show that the extent of metal-ligand interaction is key to controlling the size and uniformity in the synthesis of metal nanoclusters.). @article{nokey,
title = {Critical roles of metal–ligand complexes in the controlled synthesis of various metal nanoclusters},
author = {Ji Soo Kim and Hogeun Chang and Sungsu Kang and Seungwoo Cha and Hanguk Cho and Seung Jae Kwak and Namjun Park and Younhwa Kim and Dohun Kang and Chyan Kyung Song and Jimin Kwag and Ji-Sook Hahn and Won Bo Lee and Taeghwan Hyeon and Jungwon Park },
url = {https://www.nature.com/articles/s41467-023-38955-y.pdf},
doi = {10.1038/s41467-023-38955-y},
year = {2023},
date = {2023-06-02},
urldate = {2023-06-02},
journal = {Nature Communications},
volume = {14},
number = {3201},
abstract = {Metal nanoclusters (NCs), an important class of nanoparticles (NPs), are extremely small in size and possess quasi-molecular properties. Due to accurate stoichiometry of constituent atoms and ligands, NCs have strong structure-property relationship. The synthesis of NCs is seemingly similar to that of NPs as both are formed by colloidal phase transitions. However, they are considerably different because of metal-ligand complexes in NC synthesis. Reactive ligands can convert metal salts to complexes, actual precursors to metal NCs. During the complex formation, various metal species occur, having different reactivity and fraction depending on synthetic conditions. It can alter their degree of participation in NC synthesis and the homogeneity of final products. Herein, we investigate the effects of complex formation on the entire NC synthesis. By controlling the fraction of various Au species showing different reactivity, we find that the extent of complex formation alters reduction kinetics and the uniformity of Au NCs. We demonstrate that this concept can be universally applied to synthesize Ag, Pt, Pd, and Rh NCs.},
note = {Metal-thiolate complexes are the precursors to atomically precise metal nanoclusters. Here, the authors show that the extent of metal-ligand interaction is key to controlling the size and uniformity in the synthesis of metal nanoclusters.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Metal nanoclusters (NCs), an important class of nanoparticles (NPs), are extremely small in size and possess quasi-molecular properties. Due to accurate stoichiometry of constituent atoms and ligands, NCs have strong structure-property relationship. The synthesis of NCs is seemingly similar to that of NPs as both are formed by colloidal phase transitions. However, they are considerably different because of metal-ligand complexes in NC synthesis. Reactive ligands can convert metal salts to complexes, actual precursors to metal NCs. During the complex formation, various metal species occur, having different reactivity and fraction depending on synthetic conditions. It can alter their degree of participation in NC synthesis and the homogeneity of final products. Herein, we investigate the effects of complex formation on the entire NC synthesis. By controlling the fraction of various Au species showing different reactivity, we find that the extent of complex formation alters reduction kinetics and the uniformity of Au NCs. We demonstrate that this concept can be universally applied to synthesize Ag, Pt, Pd, and Rh NCs. |
| 62. |  | Zezhou Li, Zhiheng Xie, Yao Zhang, Xilong Mu, Jisheng Xie, Hai-Jing Yin, Ya-Wen Zhang, Colin Ophus, Jihan Zhou Probing the atomically diffuse interfaces in Pd@Pt core-shell nanoparticles in three dimensions In: Nature Communications, vol. 14, no. 2934, 2023, (Deciphering the structure and composition of bimetallic nanomaterial interfaces is key to understanding their properties. Here, the authors investigate the diffuse interfaces in Pd@Pt core-shell nanoparticles in 3D using electron tomography with atomic resolution.). @article{nokey,
title = {Probing the atomically diffuse interfaces in Pd@Pt core-shell nanoparticles in three dimensions},
author = {Zezhou Li and Zhiheng Xie and Yao Zhang and Xilong Mu and Jisheng Xie and Hai-Jing Yin and Ya-Wen Zhang and Colin Ophus and Jihan Zhou },
url = {https://www.nature.com/articles/s41467-023-38536-z.pdf},
doi = {10.1038/s41467-023-38536-z},
year = {2023},
date = {2023-05-22},
urldate = {2023-05-22},
journal = {Nature Communications},
volume = {14},
number = {2934},
abstract = {Deciphering the three-dimensional atomic structure of solid-solid interfaces in core-shell nanomaterials is the key to understand their catalytical, optical and electronic properties. Here, we probe the three-dimensional atomic structures of palladium-platinum core-shell nanoparticles at the single-atom level using atomic resolution electron tomography. We quantify the rich structural variety of core-shell nanoparticles with heteroepitaxy in 3D at atomic resolution. Instead of forming an atomically-sharp boundary, the core-shell interface is found to be atomically diffuse with an average thickness of 4.2 Å, irrespective of the particle’s morphology or crystallographic texture. The high concentration of Pd in the diffusive interface is highly related to the free Pd atoms dissolved from the Pd seeds, which is confirmed by atomic images of Pd and Pt single atoms and sub-nanometer clusters using cryogenic electron microscopy. These results advance our understanding of core-shell structures at the fundamental level, providing potential strategies into precise nanomaterial manipulation and chemical property regulation.},
note = {Deciphering the structure and composition of bimetallic nanomaterial interfaces is key to understanding their properties. Here, the authors investigate the diffuse interfaces in Pd@Pt core-shell nanoparticles in 3D using electron tomography with atomic resolution.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Deciphering the three-dimensional atomic structure of solid-solid interfaces in core-shell nanomaterials is the key to understand their catalytical, optical and electronic properties. Here, we probe the three-dimensional atomic structures of palladium-platinum core-shell nanoparticles at the single-atom level using atomic resolution electron tomography. We quantify the rich structural variety of core-shell nanoparticles with heteroepitaxy in 3D at atomic resolution. Instead of forming an atomically-sharp boundary, the core-shell interface is found to be atomically diffuse with an average thickness of 4.2 Å, irrespective of the particle’s morphology or crystallographic texture. The high concentration of Pd in the diffusive interface is highly related to the free Pd atoms dissolved from the Pd seeds, which is confirmed by atomic images of Pd and Pt single atoms and sub-nanometer clusters using cryogenic electron microscopy. These results advance our understanding of core-shell structures at the fundamental level, providing potential strategies into precise nanomaterial manipulation and chemical property regulation. |
| 61. | 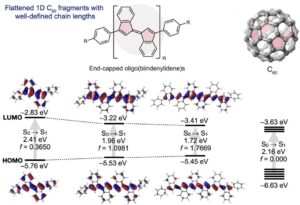 | Masahiro Hayakawa, Naoyuki Sunayama, Shu I. Takagi, Yu Matsuo, Asuka Tamaki, Shigehiro Yamaguchi, Shu Seki, Aiko Fukazawa Flattened 1D fragments of fullerene C60 that exhibit robustness toward multi-electron reduction In: Nature Communications, vol. 14, no. 2741, 2023, (Fullerenes are compelling molecular materials, but the reason for their high robustness to multi-electron reduction is still debated. Here, the authors synthesize flattened 1D fragments of fullerene C₆₀ with high robustness against multi-electron reduction.). @article{nokey,
title = {Flattened 1D fragments of fullerene C60 that exhibit robustness toward multi-electron reduction},
author = {Masahiro Hayakawa and Naoyuki Sunayama and Shu I. Takagi and Yu Matsuo and Asuka Tamaki and Shigehiro Yamaguchi and Shu Seki and Aiko Fukazawa },
url = {https://www.nature.com/articles/s41467-023-38300-3.pdf},
doi = {10.1038/s41467-023-38300-3},
year = {2023},
date = {2023-05-15},
urldate = {2023-05-15},
journal = {Nature Communications},
volume = {14},
number = {2741},
abstract = {Fullerenes are compelling molecular materials owing to their exceptional robustness toward multi-electron reduction. Although scientists have attempted to address this feature by synthesizing various fragment molecules, the origin of this electron affinity remains unclear. Several structural factors have been suggested, including high symmetry, pyramidalized carbon atoms, and five-membered ring substructures. To elucidate the role of the five-membered ring substructures without the influence of high symmetry and pyramidalized carbon atoms, we herein report the synthesis and electron-accepting properties of oligo(biindenylidene)s, a flattened one-dimensional fragment of fullerene C60. Electrochemical studies corroborated that oligo(biindenylidene)s can accept electrons up to equal to the number of five-membered rings in their main chains. Moreover, ultraviolet/visible/near-infrared absorption spectroscopy revealed that oligo(biindenylidene)s exhibit enhanced absorption covering the entire visible region relative to C60. These results highlight the significance of the pentagonal substructure for attaining stability toward multi-electron reduction and provide a strategy for the molecular design of electron-accepting π-conjugated hydrocarbons even without electron-withdrawing groups.},
note = {Fullerenes are compelling molecular materials, but the reason for their high robustness to multi-electron reduction is still debated. Here, the authors synthesize flattened 1D fragments of fullerene C₆₀ with high robustness against multi-electron reduction.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fullerenes are compelling molecular materials owing to their exceptional robustness toward multi-electron reduction. Although scientists have attempted to address this feature by synthesizing various fragment molecules, the origin of this electron affinity remains unclear. Several structural factors have been suggested, including high symmetry, pyramidalized carbon atoms, and five-membered ring substructures. To elucidate the role of the five-membered ring substructures without the influence of high symmetry and pyramidalized carbon atoms, we herein report the synthesis and electron-accepting properties of oligo(biindenylidene)s, a flattened one-dimensional fragment of fullerene C60. Electrochemical studies corroborated that oligo(biindenylidene)s can accept electrons up to equal to the number of five-membered rings in their main chains. Moreover, ultraviolet/visible/near-infrared absorption spectroscopy revealed that oligo(biindenylidene)s exhibit enhanced absorption covering the entire visible region relative to C60. These results highlight the significance of the pentagonal substructure for attaining stability toward multi-electron reduction and provide a strategy for the molecular design of electron-accepting π-conjugated hydrocarbons even without electron-withdrawing groups. |
| 60. | 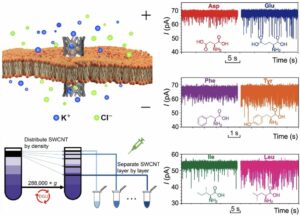 | Weichao Peng, Shuaihu Yan, Ke Zhou, Hai-Chen Wu, Lei Liu, Yuliang Zhao High-resolution discrimination of homologous and isomeric proteinogenic amino acids in nanopore sensors with ultrashort single-walled carbon nanotubes In: Nature Communications, vol. 14, no. 2662, 2023, (Ultrashort single-walled carbon nanotubes inserted in lipid bilayers can be used as nanopore sensors. Here, the authors demonstrate the high-resolution discrimination of homologous and isomeric proteinogenic amino acids with such carbon-based nanopores.). @article{nokey,
title = {High-resolution discrimination of homologous and isomeric proteinogenic amino acids in nanopore sensors with ultrashort single-walled carbon nanotubes},
author = {Weichao Peng and Shuaihu Yan and Ke Zhou and Hai-Chen Wu and Lei Liu and Yuliang Zhao },
url = {https://www.nature.com/articles/s41467-023-38399-4.pdf},
doi = {10.1038/s41467-023-38399-4},
year = {2023},
date = {2023-05-09},
urldate = {2023-05-09},
journal = {Nature Communications},
volume = {14},
number = {2662},
abstract = {The hollow and tubular structure of single-walled carbon nanotubes (SWCNTs) makes them ideal candidates for making nanopores. However, the heterogeneity of SWCNTs hinders the fabrication of robust and reproducible carbon-based nanopore sensors. Here we develop a modified density gradient ultracentrifugation approach to separate ultrashort (≈5-10 nm) SWCNTs with a narrow conductance range and construct high-resolution nanopore sensors with those tubes inserted in lipid bilayers. By conducting ionic current recordings and fluorescent imaging of Ca2+ flux through different nanopores, we prove that the ion mobilities in SWCNT nanopores are 3-5 times higher than the bulk mobility. Furthermore, we employ SWCNT nanopores to discriminate homologue or isomeric proteinogenic amino acids, which are challenging tasks for other nanopore sensors. These successes, coupled with the building of SWCNT nanopore arrays, may constitute a crucial part of the recently burgeoning protein sequencing technologies.},
note = {Ultrashort single-walled carbon nanotubes inserted in lipid bilayers can be used as nanopore sensors. Here, the authors demonstrate the high-resolution discrimination of homologous and isomeric proteinogenic amino acids with such carbon-based nanopores.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The hollow and tubular structure of single-walled carbon nanotubes (SWCNTs) makes them ideal candidates for making nanopores. However, the heterogeneity of SWCNTs hinders the fabrication of robust and reproducible carbon-based nanopore sensors. Here we develop a modified density gradient ultracentrifugation approach to separate ultrashort (≈5-10 nm) SWCNTs with a narrow conductance range and construct high-resolution nanopore sensors with those tubes inserted in lipid bilayers. By conducting ionic current recordings and fluorescent imaging of Ca2+ flux through different nanopores, we prove that the ion mobilities in SWCNT nanopores are 3-5 times higher than the bulk mobility. Furthermore, we employ SWCNT nanopores to discriminate homologue or isomeric proteinogenic amino acids, which are challenging tasks for other nanopore sensors. These successes, coupled with the building of SWCNT nanopore arrays, may constitute a crucial part of the recently burgeoning protein sequencing technologies. |
| 59. | 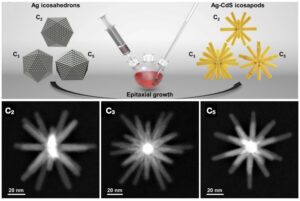 | Li Zhai, Sara T. Gebre, Bo Chen, Dan Xu, Junze Chen, Zijian Li, Yawei Liu, Hua Yang, Chongyi Ling, Yiyao Ge, Wei Zhai, Changsheng Chen, Lu Ma, Qinghua Zhang, Xuefei Li, Yujie Yan, Xinyu Huang, Lujiang Li, Zhiqiang Guan, Chen-Lei Tao, Zhiqi Huang, Hongyi Wang, Jinze Liang, Ye Zhu, Chun-Sing Lee, Peng Wang, Chunfeng Zhang, Lin Gu, Yonghua Du, Tianquan Lian, Hua Zhang, Xue-Jun Wu Epitaxial growth of highly symmetrical branched noble metal-semiconductor heterostructures with efficient plasmon-induced hot-electron transfer In: Nature Communications, vol. 14, no. 2538, 2023, (Epitaxial growth of heterostructures composed of materials with large lattice mismatch is challenging. Here, the authors reported the epitaxy of II-VI semiconductor nanorods on plasmonic noble metal, despite a lattice mismatch of more than 40%.). @article{nokey,
title = {Epitaxial growth of highly symmetrical branched noble metal-semiconductor heterostructures with efficient plasmon-induced hot-electron transfer},
author = {Li Zhai and Sara T. Gebre and Bo Chen and Dan Xu and Junze Chen and Zijian Li and Yawei Liu and Hua Yang and Chongyi Ling and Yiyao Ge and Wei Zhai and Changsheng Chen and Lu Ma and Qinghua Zhang and Xuefei Li and Yujie Yan and Xinyu Huang and Lujiang Li and Zhiqiang Guan and Chen-Lei Tao and Zhiqi Huang and Hongyi Wang and Jinze Liang and Ye Zhu and Chun-Sing Lee and Peng Wang and Chunfeng Zhang and Lin Gu and Yonghua Du and Tianquan Lian and Hua Zhang and Xue-Jun Wu },
url = {https://www.nature.com/articles/s41467-023-38237-7.pdf},
doi = {10.1038/s41467-023-38237-7},
year = {2023},
date = {2023-05-03},
urldate = {2023-05-03},
journal = {Nature Communications},
volume = {14},
number = {2538},
abstract = {Epitaxial growth is one of the most commonly used strategies to precisely tailor heterostructures with well-defined compositions, morphologies, crystal phases, and interfaces for various applications. However, as epitaxial growth requires a small interfacial lattice mismatch between the components, it remains a challenge for the epitaxial synthesis of heterostructures constructed by materials with large lattice mismatch and/or different chemical bonding, especially the noble metal-semiconductor heterostructures. Here, we develop a noble metal-seeded epitaxial growth strategy to prepare highly symmetrical noble metal-semiconductor branched heterostructures with desired spatial configurations, i.e., twenty CdS (or CdSe) nanorods epitaxially grown on twenty exposed (111) facets of Ag icosahedral nanocrystal, albeit a large lattice mismatch (more than 40%). Importantly, a high quantum yield (QY) of plasmon-induced hot-electron transferred from Ag to CdS was observed in epitaxial Ag-CdS icosapods (18.1%). This work demonstrates that epitaxial growth can be achieved in heterostructures composed of materials with large lattice mismatches. The constructed epitaxial noble metal-semiconductor interfaces could be an ideal platform for investigating the role of interfaces in various physicochemical processes.},
note = {Epitaxial growth of heterostructures composed of materials with large lattice mismatch is challenging. Here, the authors reported the epitaxy of II-VI semiconductor nanorods on plasmonic noble metal, despite a lattice mismatch of more than 40%.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Epitaxial growth is one of the most commonly used strategies to precisely tailor heterostructures with well-defined compositions, morphologies, crystal phases, and interfaces for various applications. However, as epitaxial growth requires a small interfacial lattice mismatch between the components, it remains a challenge for the epitaxial synthesis of heterostructures constructed by materials with large lattice mismatch and/or different chemical bonding, especially the noble metal-semiconductor heterostructures. Here, we develop a noble metal-seeded epitaxial growth strategy to prepare highly symmetrical noble metal-semiconductor branched heterostructures with desired spatial configurations, i.e., twenty CdS (or CdSe) nanorods epitaxially grown on twenty exposed (111) facets of Ag icosahedral nanocrystal, albeit a large lattice mismatch (more than 40%). Importantly, a high quantum yield (QY) of plasmon-induced hot-electron transferred from Ag to CdS was observed in epitaxial Ag-CdS icosapods (18.1%). This work demonstrates that epitaxial growth can be achieved in heterostructures composed of materials with large lattice mismatches. The constructed epitaxial noble metal-semiconductor interfaces could be an ideal platform for investigating the role of interfaces in various physicochemical processes. |
| 58. | 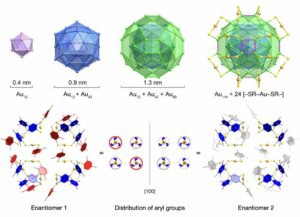 | Li-Juan Liu, Fahri Alkan, Shengli Zhuang, Dongyi Liu, Tehseen Nawaz, Jun Guo, Xiaozhou Luo, Jian He Atomically precise gold nanoclusters at the molecular-to-metallic transition with intrinsic chirality from surface layers In: Nature Communications, vol. 14, no. 2397, 2023, (Chiral metal nanoclusters prepared from achiral ligands generally contain chiral kernel structures. Here, the authors report an alternative type of gold nanoclusters whose intrinsic chirality arises solely from the arrangement of the organic components on their surface.). @article{nokey,
title = {Atomically precise gold nanoclusters at the molecular-to-metallic transition with intrinsic chirality from surface layers},
author = {Li-Juan Liu and Fahri Alkan and Shengli Zhuang and Dongyi Liu and Tehseen Nawaz and Jun Guo and Xiaozhou Luo and Jian He},
url = {https://www.nature.com/articles/s41467-023-38179-0.pdf},
doi = {10.1038/s41467-023-38179-0},
year = {2023},
date = {2023-04-26},
urldate = {2023-04-26},
journal = {Nature Communications},
volume = {14},
number = {2397},
abstract = {The advances in determining the total structure of atomically precise metal nanoclusters have prompted extensive exploration into the origins of chirality in nanoscale systems. While chirality is generally transferrable from the surface layer to the metal–ligand interface and kernel, we present here an alternative type of gold nanoclusters (138 gold core atoms with 48 2,4-dimethylbenzenethiolate surface ligands) whose inner structures are not asymmetrically induced by chiral patterns of the outermost aromatic substituents. This phenomenon can be explained by the highly dynamic behaviors of aromatic rings in the thiolates assembled via π − π stacking and C − H···π interactions. In addition to being a thiolate-protected nanocluster with uncoordinated surface gold atoms, the reported Au138 motif expands the size range of gold nanoclusters having both molecular and metallic properties. Our current work introduces an important class of nanoclusters with intrinsic chirality from surface layers rather than inner structures and will aid in elucidating the transition of gold nanoclusters from their molecular to metallic states.},
note = {Chiral metal nanoclusters prepared from achiral ligands generally contain chiral kernel structures. Here, the authors report an alternative type of gold nanoclusters whose intrinsic chirality arises solely from the arrangement of the organic components on their surface.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The advances in determining the total structure of atomically precise metal nanoclusters have prompted extensive exploration into the origins of chirality in nanoscale systems. While chirality is generally transferrable from the surface layer to the metal–ligand interface and kernel, we present here an alternative type of gold nanoclusters (138 gold core atoms with 48 2,4-dimethylbenzenethiolate surface ligands) whose inner structures are not asymmetrically induced by chiral patterns of the outermost aromatic substituents. This phenomenon can be explained by the highly dynamic behaviors of aromatic rings in the thiolates assembled via π − π stacking and C − H···π interactions. In addition to being a thiolate-protected nanocluster with uncoordinated surface gold atoms, the reported Au138 motif expands the size range of gold nanoclusters having both molecular and metallic properties. Our current work introduces an important class of nanoclusters with intrinsic chirality from surface layers rather than inner structures and will aid in elucidating the transition of gold nanoclusters from their molecular to metallic states. |
| 57. | 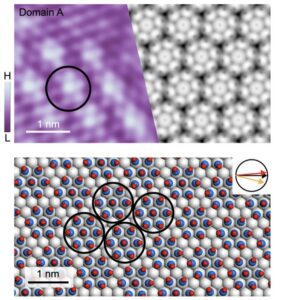 | Huiru Liu, Heping Li, Yu He, Peng Cheng, Yi-Qi Zhang, Baojie Feng, Hui Li, Kehui Wu, Lan Chen Condensation and asymmetric amplification of chirality in achiral molecules adsorbed on an achiral surface In: Nature Communications, vol. 14, no. 2100, 2023, (The origin of homochirality in nature is an important but open question. Here, the authors provide insight into the physicochemical origin of homochirality through surface adsorption on the model of adlayers of achiral carbon monoxide molecules on an achiral Au(111) surface.). @article{nokey,
title = {Condensation and asymmetric amplification of chirality in achiral molecules adsorbed on an achiral surface},
author = {Huiru Liu and Heping Li and Yu He and Peng Cheng and Yi-Qi Zhang and Baojie Feng and Hui Li and Kehui Wu and Lan Chen },
url = {https://www.nature.com/articles/s41467-023-37904-z.pdf},
doi = {10.1038/s41467-023-37904-z},
year = {2023},
date = {2023-04-13},
urldate = {2023-04-13},
journal = {Nature Communications},
volume = {14},
number = {2100},
abstract = {The origin of homochirality in nature is an important but open question. Here, we demonstrate a simple organizational chiral system constructed by achiral carbon monoxide (CO) molecules adsorbed on an achiral Au(111) substrate. Combining scanning tunneling microscope (STM) measurements with density-functional-theory (DFT) calculations, two dissymmetric cluster phases consisting of chiral CO heptamers are revealed. By applied high bias voltage, the stable racemic cluster phase can be transformed into a metastable uniform phase consisting of CO monomers. Further, during the recondensation of a cluster phase after lowering down bias voltage, an enantiomeric excess and its chiral amplification occur, resulting in a homochirality. Such asymmetry amplification is found to be both kinetically feasible and thermodynamically favorable. Our observations provide insight into the physicochemical origin of homochirality through surface adsorption and suggest a general phenomenon that can influence enantioselective chemical processes such as chiral separations and heterogeneous asymmetric catalysis.},
note = {The origin of homochirality in nature is an important but open question. Here, the authors provide insight into the physicochemical origin of homochirality through surface adsorption on the model of adlayers of achiral carbon monoxide molecules on an achiral Au(111) surface.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The origin of homochirality in nature is an important but open question. Here, we demonstrate a simple organizational chiral system constructed by achiral carbon monoxide (CO) molecules adsorbed on an achiral Au(111) substrate. Combining scanning tunneling microscope (STM) measurements with density-functional-theory (DFT) calculations, two dissymmetric cluster phases consisting of chiral CO heptamers are revealed. By applied high bias voltage, the stable racemic cluster phase can be transformed into a metastable uniform phase consisting of CO monomers. Further, during the recondensation of a cluster phase after lowering down bias voltage, an enantiomeric excess and its chiral amplification occur, resulting in a homochirality. Such asymmetry amplification is found to be both kinetically feasible and thermodynamically favorable. Our observations provide insight into the physicochemical origin of homochirality through surface adsorption and suggest a general phenomenon that can influence enantioselective chemical processes such as chiral separations and heterogeneous asymmetric catalysis. |
| 56. | 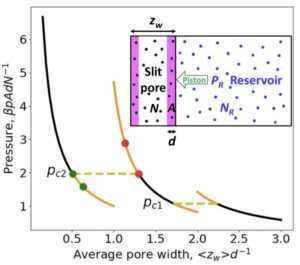 | Wei Dong Nanoscale thermodynamics needs the concept of a disjoining chemical potential In: Nature Communications, vol. 14, no. 1824, 2023, (Matter behaves differently at the nanoscale. Here, the author introduces the concept of a disjoining chemical potential for nanoscale thermodynamics, showing that thermodynamic functions depend on the environment, and suggests possible experimental verifications.). @article{nokey,
title = {Nanoscale thermodynamics needs the concept of a disjoining chemical potential},
author = {Wei Dong},
url = {https://www.nature.com/articles/s41467-023-36970-7.pdf},
doi = {10.1038/s41467-023-36970-7},
year = {2023},
date = {2023-04-01},
urldate = {2023-04-01},
journal = {Nature Communications},
volume = {14},
number = {1824},
abstract = {Disjoining pressure was discovered by Derjaguin in 1930’s, which describes the difference between the pressure of a strongly confined fluid and the corresponding one in a bulk phase. It has been revealed recently that the disjoining pressure is at the origin of distinct differential and integral surface tensions for strongly confined fluids. Here we show how the twin concept, disjoining chemical potential, arises in a reminiscent way although it comes out eighty years later. This twin concept advances our understanding of nanoscale thermodynamics. Ensemble-dependence (or environment-dependence) is one hallmark of thermodynamics of small systems. We show that integral surface tension is ensemble-dependent while differential surface tension is not. Moreover, two generalized Gibbs-Duhem equations involving integral surface tensions are derived, as well as two additional adsorption equations relating surface tensions to adsorption-induced strains. All the results obtained in this work further evidence that an approach alternative of Hill’s nanothermodynamics is possible, by extending Gibbs surface thermodynamics instead of resorting to Hill’s replica trick. Moreover, we find a compression-expansion hysteresis without any underlying phase transition.},
note = {Matter behaves differently at the nanoscale. Here, the author introduces the concept of a disjoining chemical potential for nanoscale thermodynamics, showing that thermodynamic functions depend on the environment, and suggests possible experimental verifications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Disjoining pressure was discovered by Derjaguin in 1930’s, which describes the difference between the pressure of a strongly confined fluid and the corresponding one in a bulk phase. It has been revealed recently that the disjoining pressure is at the origin of distinct differential and integral surface tensions for strongly confined fluids. Here we show how the twin concept, disjoining chemical potential, arises in a reminiscent way although it comes out eighty years later. This twin concept advances our understanding of nanoscale thermodynamics. Ensemble-dependence (or environment-dependence) is one hallmark of thermodynamics of small systems. We show that integral surface tension is ensemble-dependent while differential surface tension is not. Moreover, two generalized Gibbs-Duhem equations involving integral surface tensions are derived, as well as two additional adsorption equations relating surface tensions to adsorption-induced strains. All the results obtained in this work further evidence that an approach alternative of Hill’s nanothermodynamics is possible, by extending Gibbs surface thermodynamics instead of resorting to Hill’s replica trick. Moreover, we find a compression-expansion hysteresis without any underlying phase transition. |
| 55. |  | Eleonora Calì, Melonie P. Thomas, Rama Vasudevan, Ji Wu, Oriol Gavalda-Diaz, Katharina Marquardt, Eduardo Saiz, Dragos Neagu, Raymond R. Unocic, Stephen C. Parker, Beth S. Guiton, David J. Payne Real-time insight into the multistage mechanism of nanoparticle exsolution from a perovskite host surface In: Nature Communications, vol. 14, no. 1754, 2023, (The separation of nanoparticles from oxide hosts by exsolution forms the basis for catalytic and energy-related applications. Here, the authors elucidate the multistep mechanism of exsolution at perovskite surfaces by combining real-time electron microscopy and computational methods.). @article{nokey,
title = {Real-time insight into the multistage mechanism of nanoparticle exsolution from a perovskite host surface},
author = {Eleonora Calì and Melonie P. Thomas and Rama Vasudevan and Ji Wu and Oriol Gavalda-Diaz and Katharina Marquardt and Eduardo Saiz and Dragos Neagu and Raymond R. Unocic and Stephen C. Parker and Beth S. Guiton and David J. Payne },
url = {https://www.nature.com/articles/s41467-023-37212-6.pdf},
doi = {10.1038/s41467-023-37212-6},
year = {2023},
date = {2023-03-29},
urldate = {2023-03-29},
journal = {Nature Communications},
volume = {14},
number = {1754},
abstract = {In exsolution, nanoparticles form by emerging from oxide hosts by application of redox driving forces, leading to transformative advances in stability, activity, and efficiency over deposition techniques, and resulting in a wide range of new opportunities for catalytic, energy and net-zero-related technologies. However, the mechanism of exsolved nanoparticle nucleation and perovskite structural evolution, has, to date, remained unclear. Herein, we shed light on this elusive process by following in real time Ir nanoparticle emergence from a SrTiO3 host oxide lattice, using in situ high-resolution electron microscopy in combination with computational simulations and machine learning analytics. We show that nucleation occurs via atom clustering, in tandem with host evolution, revealing the participation of surface defects and host lattice restructuring in trapping Ir atoms to initiate nanoparticle formation and growth. These insights provide a theoretical platform and practical recommendations to further the development of highly functional and broadly applicable exsolvable materials.},
note = {The separation of nanoparticles from oxide hosts by exsolution forms the basis for catalytic and energy-related applications. Here, the authors elucidate the multistep mechanism of exsolution at perovskite surfaces by combining real-time electron microscopy and computational methods.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In exsolution, nanoparticles form by emerging from oxide hosts by application of redox driving forces, leading to transformative advances in stability, activity, and efficiency over deposition techniques, and resulting in a wide range of new opportunities for catalytic, energy and net-zero-related technologies. However, the mechanism of exsolved nanoparticle nucleation and perovskite structural evolution, has, to date, remained unclear. Herein, we shed light on this elusive process by following in real time Ir nanoparticle emergence from a SrTiO3 host oxide lattice, using in situ high-resolution electron microscopy in combination with computational simulations and machine learning analytics. We show that nucleation occurs via atom clustering, in tandem with host evolution, revealing the participation of surface defects and host lattice restructuring in trapping Ir atoms to initiate nanoparticle formation and growth. These insights provide a theoretical platform and practical recommendations to further the development of highly functional and broadly applicable exsolvable materials. |
| 54. |  | Mo Xie, Weina Fang, Zhibei Qu, Yang Hu, Yichi Zhang, Jie Chao, Jiye Shi, Lihua Wang, Lianhui Wang, Yang Tian, Chunhai Fan, Huajie Liu
High-entropy alloy nanopatterns by prescribed metallization of DNA origami templates In: Nature Communications, vol. 14, no. 1745, 2023, (Morphology, composition, and uniformity of highly entropic nanoalloys are critical to their properties and applications. Here, the authors develop a DNA origami-based metallization reaction concept for the precise synthesis of multimetallic nanopatterns.). @article{nokey,
title = {High-entropy alloy nanopatterns by prescribed metallization of DNA origami templates},
author = {Mo Xie and Weina Fang and Zhibei Qu and Yang Hu and Yichi Zhang and Jie Chao and Jiye Shi and Lihua Wang and Lianhui Wang and Yang Tian and Chunhai Fan and Huajie Liu
},
url = {https://www.nature.com/articles/s41467-023-37333-y.pdf},
doi = {10.1038/s41467-023-37333-y},
year = {2023},
date = {2023-03-28},
urldate = {2023-03-28},
journal = {Nature Communications},
volume = {14},
number = {1745},
abstract = {High-entropy multimetallic nanopatterns with controlled morphology, composition and uniformity hold great potential for developing nanoelectronics, nanophotonics and catalysis. Nevertheless, the lack of general methods for patterning multiple metals poses a limit. Here, we develop a DNA origami-based metallization reaction system to prescribe multimetallic nanopatterns with peroxidase-like activities. We find that strong coordination between metal elements and DNA bases enables the accumulation of metal ions on protruding clustered DNA (pcDNA) that are prescribed on DNA origami. As a result of the condensation of pcDNA, these sites can serve as nucleation site for metal plating. We have synthesized multimetallic nanopatterns composed of up to five metal elements (Co, Pd, Pt, Ag and Ni), and obtained insights on elemental uniformity control at the nanoscale. This method provides an alternative pathway to construct a library of multimetallic nanopatterns.},
note = {Morphology, composition, and uniformity of highly entropic nanoalloys are critical to their properties and applications. Here, the authors develop a DNA origami-based metallization reaction concept for the precise synthesis of multimetallic nanopatterns.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
High-entropy multimetallic nanopatterns with controlled morphology, composition and uniformity hold great potential for developing nanoelectronics, nanophotonics and catalysis. Nevertheless, the lack of general methods for patterning multiple metals poses a limit. Here, we develop a DNA origami-based metallization reaction system to prescribe multimetallic nanopatterns with peroxidase-like activities. We find that strong coordination between metal elements and DNA bases enables the accumulation of metal ions on protruding clustered DNA (pcDNA) that are prescribed on DNA origami. As a result of the condensation of pcDNA, these sites can serve as nucleation site for metal plating. We have synthesized multimetallic nanopatterns composed of up to five metal elements (Co, Pd, Pt, Ag and Ni), and obtained insights on elemental uniformity control at the nanoscale. This method provides an alternative pathway to construct a library of multimetallic nanopatterns. |
| 53. | 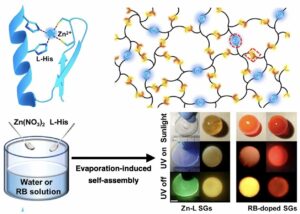 | Fei Nie, Ke-Zhi Wang, Dongpeng Yan Supramolecular glasses with color-tunable circularly polarized afterglow through evaporation-induced self-assembly of chiral metal–organic complexes In: Nature Communications, vol. 14, no. 1654, 2023, (Material designs with multicolor circularly polarized emissions are desirable for photonic applications. Here, the authors report supramolecular glasses based on self-assembled chiral metal–organic complexes with color-tunable circularly polarized afterglow.). @article{nokey,
title = {Supramolecular glasses with color-tunable circularly polarized afterglow through evaporation-induced self-assembly of chiral metal–organic complexes},
author = {Fei Nie and Ke-Zhi Wang and Dongpeng Yan },
url = {https://www.nature.com/articles/s41467-023-37331-0.pdf},
doi = {10.1038/s41467-023-37331-0},
year = {2023},
date = {2023-03-24},
urldate = {2023-03-24},
journal = {Nature Communications},
volume = {14},
number = {1654},
abstract = {The fabrication of chiral molecules into macroscopic systems has many valuable applications, especially in the fields of optical displays, data encryption, information storage, and so on. Here, we design and prepare a serious of supramolecular glasses (SGs) based on Zn-L-Histidine complexes, via an evaporation-induced self-assembly (EISA) strategy. Metal-ligand interactions between the zinc(II) ion and chiral L-Histidine endow the SGs with interesting circularly polarized afterglow (CPA). Multicolored CPA emissions from blue to red with dissymmetry factor as high as 9.5 × 10−3 and excited-state lifetime up to 356.7 ms are achieved under ambient conditions. Therefore, this work not only communicates the bulk SGs with wide-tunable afterglow and large circular polarization, but also provides an EISA method for the macroscopic self-assembly of chiral metal–organic hybrids toward photonic applications.
},
note = {Material designs with multicolor circularly polarized emissions are desirable for photonic applications. Here, the authors report supramolecular glasses based on self-assembled chiral metal–organic complexes with color-tunable circularly polarized afterglow.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The fabrication of chiral molecules into macroscopic systems has many valuable applications, especially in the fields of optical displays, data encryption, information storage, and so on. Here, we design and prepare a serious of supramolecular glasses (SGs) based on Zn-L-Histidine complexes, via an evaporation-induced self-assembly (EISA) strategy. Metal-ligand interactions between the zinc(II) ion and chiral L-Histidine endow the SGs with interesting circularly polarized afterglow (CPA). Multicolored CPA emissions from blue to red with dissymmetry factor as high as 9.5 × 10−3 and excited-state lifetime up to 356.7 ms are achieved under ambient conditions. Therefore, this work not only communicates the bulk SGs with wide-tunable afterglow and large circular polarization, but also provides an EISA method for the macroscopic self-assembly of chiral metal–organic hybrids toward photonic applications.
|
| 52. |  | Maximilian Dreher, Pierre Martin Dombrowski, Matthias Wolfgang Tripp, Niels Münster, Ulrich Koert, Gregor Witte Shape control in 2D molecular nanosheets by tuning anisotropic intermolecular interactions and assembly kinetics In: Nature Communications, vol. 14, no. 1554, 2023, (Structuring organic films is of scientific and technological interest. Here, the authors use partially fluorinated organic molecules exhibiting strong intermolecular interactions to form extended 2D molecular nanosheets and control their shape through growth and desorption kinetics.). @article{nokey,
title = {Shape control in 2D molecular nanosheets by tuning anisotropic intermolecular interactions and assembly kinetics},
author = {Maximilian Dreher and Pierre Martin Dombrowski and Matthias Wolfgang Tripp and Niels Münster and Ulrich Koert and Gregor Witte },
url = {https://www.nature.com/articles/s41467-023-37203-7.pdf},
doi = {10.1038/s41467-023-37203-7},
year = {2023},
date = {2023-03-21},
urldate = {2023-03-21},
journal = {Nature Communications},
volume = {14},
number = {1554},
abstract = {Since molecular materials often decompose upon exposure to radiation, lithographic patterning techniques established for inorganic materials are usually not applicable for the fabrication of organic nanostructures. Instead, molecular self-organisation must be utilised to achieve bottom-up growth of desired structures. Here, we demonstrate control over the mesoscopic shape of 2D molecular nanosheets without affecting their nanoscopic molecular packing motif, using molecules that do not form lateral covalent bonds. We show that anisotropic attractive Coulomb forces between partially fluorinated pentacenes lead to the growth of distinctly elongated nanosheets and that the direction of elongation differs between nanosheets that were grown and ones that were fabricated by partial desorption of a complete molecular monolayer. Using kinetic Monte Carlo simulations, we show that lateral intermolecular interactions alone are sufficient to rationalise the different kinetics of structure formation during nanosheet growth and desorption, without inclusion of interactions between the molecules and the supporting MoS2 substrate. By comparison of the behaviour of differently fluorinated molecules, experimentally and computationally, we can identify properties of molecules with regard to interactions and molecular packing motifs that are required for an effective utilisation of the observed effect.},
note = {Structuring organic films is of scientific and technological interest. Here, the authors use partially fluorinated organic molecules exhibiting strong intermolecular interactions to form extended 2D molecular nanosheets and control their shape through growth and desorption kinetics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Since molecular materials often decompose upon exposure to radiation, lithographic patterning techniques established for inorganic materials are usually not applicable for the fabrication of organic nanostructures. Instead, molecular self-organisation must be utilised to achieve bottom-up growth of desired structures. Here, we demonstrate control over the mesoscopic shape of 2D molecular nanosheets without affecting their nanoscopic molecular packing motif, using molecules that do not form lateral covalent bonds. We show that anisotropic attractive Coulomb forces between partially fluorinated pentacenes lead to the growth of distinctly elongated nanosheets and that the direction of elongation differs between nanosheets that were grown and ones that were fabricated by partial desorption of a complete molecular monolayer. Using kinetic Monte Carlo simulations, we show that lateral intermolecular interactions alone are sufficient to rationalise the different kinetics of structure formation during nanosheet growth and desorption, without inclusion of interactions between the molecules and the supporting MoS2 substrate. By comparison of the behaviour of differently fluorinated molecules, experimentally and computationally, we can identify properties of molecules with regard to interactions and molecular packing motifs that are required for an effective utilisation of the observed effect. |
| 51. | 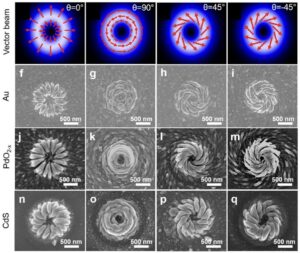 | Xiaolin Lu, Xujie Wang, Shuangshuang Wang, Tao Ding Polarization-directed growth of spiral nanostructures by laser direct writing with vector beams In: Nature Communications, vol. 14, no. 1422, 2023, (Chiral nanostructures are in demand for various applications, but large-scale fabrication is a technical challenge. Here, the authors report polarization-directed chiral growth of complex spiral patterns by laser direct writing with vector beams.). @article{nokey,
title = {Polarization-directed growth of spiral nanostructures by laser direct writing with vector beams},
author = {Xiaolin Lu and Xujie Wang and Shuangshuang Wang and Tao Ding },
url = {https://www.nature.com/articles/s41467-023-37048-0.pdf},
doi = {10.1038/s41467-023-37048-0},
year = {2023},
date = {2023-03-14},
urldate = {2023-03-14},
journal = {Nature Communications},
volume = {14},
number = {1422},
abstract = {Chirality is pivotal in nature which attracts wide research interests from all disciplines and creating chiral matter is one of the central themes for chemists and material scientists. Despite of significant efforts, a simple, cost-effective and general method that can produce different kinds of chiral metamaterials with high regularity and tailorability is still demanding but greatly missing. Here, we introduce polarization-directed growth of spiral nanostructures via vector beams, which is simple, tailorable and generally applicable to both plasmonic and dielectric materials. The self-aligned near field enhances the photochemical growth along the polarization, which is crucial for the oriented growth. The obtained plasmonic chiral nanostructures present prominent optical activity with a g-factor up to 0.4, which can be tuned by adjusting the spirality of the vector beams. These spiral plasmonic nanostructures can be used for the sensing of different chiral enantiomers. The dielectric chiral metasurfaces can also be formed in arrays of sub-mm scale, which exhibit a g-factor over 0.1. However, photoluminescence of chiral cadmium sulfide presents a very weak luminescence g-factor with the excitation of linearly polarized light. A number of applications can be envisioned with these chiral nanostructures such as chiral sensing, chiral separation and chiral information storage.},
note = {Chiral nanostructures are in demand for various applications, but large-scale fabrication is a technical challenge. Here, the authors report polarization-directed chiral growth of complex spiral patterns by laser direct writing with vector beams.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chirality is pivotal in nature which attracts wide research interests from all disciplines and creating chiral matter is one of the central themes for chemists and material scientists. Despite of significant efforts, a simple, cost-effective and general method that can produce different kinds of chiral metamaterials with high regularity and tailorability is still demanding but greatly missing. Here, we introduce polarization-directed growth of spiral nanostructures via vector beams, which is simple, tailorable and generally applicable to both plasmonic and dielectric materials. The self-aligned near field enhances the photochemical growth along the polarization, which is crucial for the oriented growth. The obtained plasmonic chiral nanostructures present prominent optical activity with a g-factor up to 0.4, which can be tuned by adjusting the spirality of the vector beams. These spiral plasmonic nanostructures can be used for the sensing of different chiral enantiomers. The dielectric chiral metasurfaces can also be formed in arrays of sub-mm scale, which exhibit a g-factor over 0.1. However, photoluminescence of chiral cadmium sulfide presents a very weak luminescence g-factor with the excitation of linearly polarized light. A number of applications can be envisioned with these chiral nanostructures such as chiral sensing, chiral separation and chiral information storage. |
| 50. | 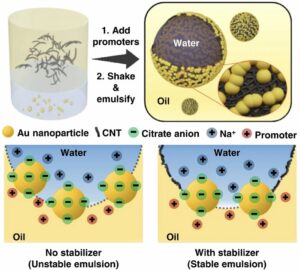 | Yingrui Zhang, Ziwei Ye, Chunchun Li, Qinglu Chen, Wafaa Aljuhani, Yiming Huang, Xin Xu, Chunfei Wu, Steven E. J. Bell, Yikai Xu General approach to surface-accessible plasmonic Pickering emulsions for SERS sensing and interfacial catalysis In: Nature Communications, vol. 14, no. 1392, 2023, (The preparation of Pickering emulsions from nanoparticles commonly requires modification of the particles’ surface chemistry. Here, the authors demonstrate a chemical modifier-free approach for the preparation of long-term stable Pickering emulsions for SERS sensing and catalytic applications.). @article{nokey,
title = {General approach to surface-accessible plasmonic Pickering emulsions for SERS sensing and interfacial catalysis},
author = {Yingrui Zhang and Ziwei Ye and Chunchun Li and Qinglu Chen and Wafaa Aljuhani and Yiming Huang and Xin Xu and Chunfei Wu and Steven E. J. Bell and Yikai Xu },
url = {https://www.nature.com/articles/s41467-023-37001-1.pdf},
doi = {10.1038/s41467-023-37001-1},
year = {2023},
date = {2023-03-13},
urldate = {2023-03-13},
journal = {Nature Communications},
volume = {14},
number = {1392},
abstract = {Pickering emulsions represent an important class of functional materials with potential applications in sustainability and healthcare. Currently, the synthesis of Pickering emulsions relies heavily on the use of strongly adsorbing molecular modifiers to tune the surface chemistry of the nanoparticle constituents. This approach is inconvenient and potentially a dead-end for many applications since the adsorbed modifiers prevent interactions between the functional nanosurface and its surroundings. Here, we demonstrate a general modifier-free approach to construct Pickering emulsions by using a combination of stabilizer particles, which stabilize the emulsion droplet, and a second population of unmodified functional particles that sit alongside the stabilizers at the interface. Freeing Pickering emulsions from chemical modifiers unlocks their potential across a range of applications including plasmonic sensing and interfacial catalysis that have previously been challenging to achieve. More broadly, this strategy provides an approach to the development of surface-accessible nanomaterials with enhanced and/or additional properties from a wide range of nano-building blocks including organic nanocrystals, carbonaceous materials, metals and oxides.},
note = {The preparation of Pickering emulsions from nanoparticles commonly requires modification of the particles’ surface chemistry. Here, the authors demonstrate a chemical modifier-free approach for the preparation of long-term stable Pickering emulsions for SERS sensing and catalytic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pickering emulsions represent an important class of functional materials with potential applications in sustainability and healthcare. Currently, the synthesis of Pickering emulsions relies heavily on the use of strongly adsorbing molecular modifiers to tune the surface chemistry of the nanoparticle constituents. This approach is inconvenient and potentially a dead-end for many applications since the adsorbed modifiers prevent interactions between the functional nanosurface and its surroundings. Here, we demonstrate a general modifier-free approach to construct Pickering emulsions by using a combination of stabilizer particles, which stabilize the emulsion droplet, and a second population of unmodified functional particles that sit alongside the stabilizers at the interface. Freeing Pickering emulsions from chemical modifiers unlocks their potential across a range of applications including plasmonic sensing and interfacial catalysis that have previously been challenging to achieve. More broadly, this strategy provides an approach to the development of surface-accessible nanomaterials with enhanced and/or additional properties from a wide range of nano-building blocks including organic nanocrystals, carbonaceous materials, metals and oxides. |
| 49. |  | Nan Cao, Biao Yang, Alexander Riss, Johanna Rosen, Jonas Björk, Johannes V. Barth
On-surface synthesis of enetriynes In: Nature Communications, vol. 14, no. 1255, 2023, (Enetriynes, which belong to the enyne family, are characterized by a distinct electron-rich carbon-bonding scheme. Here, the authors report the formation of enetriynes with high selectivity by tetramerization of terminal alkynes on Ag(100).). @article{nokey,
title = {On-surface synthesis of enetriynes},
author = {Nan Cao and Biao Yang and Alexander Riss and Johanna Rosen and Jonas Björk and Johannes V. Barth
},
url = {https://www.nature.com/articles/s41467-023-36828-y.pdf},
doi = {10.1038/s41467-023-36828-y},
year = {2023},
date = {2023-03-06},
urldate = {2023-03-06},
journal = {Nature Communications},
volume = {14},
number = {1255},
abstract = {Belonging to the enyne family, enetriynes comprise a distinct electron-rich all-carbon bonding scheme. However, the lack of convenient synthesis protocols limits the associated application potential within, e.g., biochemistry and materials science. Herein we introduce a pathway for highly selective enetriyne formation via tetramerization of terminal alkynes on a Ag(100) surface. Taking advantage of a directing hydroxyl group, we steer molecular assembly and reaction processes on square lattices. Induced by O2 exposure the terminal alkyne moieties deprotonate and organometallic bis-acetylide dimer arrays evolve. Upon subsequent thermal annealing tetrameric enetriyne-bridged compounds are generated in high yield, readily self-assembling into regular networks. We combine high-resolution scanning probe microscopy, X-ray photoelectron spectroscopy and density functional theory calculations to examine the structural features, bonding characteristics and the underlying reaction mechanism. Our study introduces an integrated strategy for the precise fabrication of functional enetriyne species, thus providing access to a distinct class of highly conjugated π-system compounds.},
note = {Enetriynes, which belong to the enyne family, are characterized by a distinct electron-rich carbon-bonding scheme. Here, the authors report the formation of enetriynes with high selectivity by tetramerization of terminal alkynes on Ag(100).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Belonging to the enyne family, enetriynes comprise a distinct electron-rich all-carbon bonding scheme. However, the lack of convenient synthesis protocols limits the associated application potential within, e.g., biochemistry and materials science. Herein we introduce a pathway for highly selective enetriyne formation via tetramerization of terminal alkynes on a Ag(100) surface. Taking advantage of a directing hydroxyl group, we steer molecular assembly and reaction processes on square lattices. Induced by O2 exposure the terminal alkyne moieties deprotonate and organometallic bis-acetylide dimer arrays evolve. Upon subsequent thermal annealing tetrameric enetriyne-bridged compounds are generated in high yield, readily self-assembling into regular networks. We combine high-resolution scanning probe microscopy, X-ray photoelectron spectroscopy and density functional theory calculations to examine the structural features, bonding characteristics and the underlying reaction mechanism. Our study introduces an integrated strategy for the precise fabrication of functional enetriyne species, thus providing access to a distinct class of highly conjugated π-system compounds. |
| 48. |  | Hongwei Cheng, Xiaoxia Zhu, Xiaowei Cheng, Pengzhan Cai, Jie Liu, Huijun Yao, Ling Zhang, Jinglai Duan Mechanical metamaterials made of freestanding quasi-BCC nanolattices of gold and copper with ultra-high energy absorption capacity In: Nature Communications, vol. 14, no. 1243, 2023, (The fabrication of freestanding 3D lattice structures with beam diameters less than 100 nm is a considerable challenge. Here, the authors report quasi-BCC nanolattices of gold and copper, featuring beam diameters as low as 34 nm, that demonstrate an exceptionally high capacity for energy absorption.). @article{nokey,
title = {Mechanical metamaterials made of freestanding quasi-BCC nanolattices of gold and copper with ultra-high energy absorption capacity},
author = {Hongwei Cheng and Xiaoxia Zhu and Xiaowei Cheng and Pengzhan Cai and Jie Liu and Huijun Yao and Ling Zhang and Jinglai Duan },
url = {https://www.nature.com/articles/s41467-023-36965-4.pdf},
doi = {10.1038/s41467-023-36965-4},
year = {2023},
date = {2023-03-04},
urldate = {2023-03-04},
journal = {Nature Communications},
volume = {14},
number = {1243},
abstract = {Nanolattices exhibit attractive mechanical properties such as high strength, high specific strength, and high energy absorption. However, at present, such materials cannot achieve effective fusion of the above properties and scalable production, which hinders their applications in energy conversion and other fields. Herein, we report gold and copper quasi-body centered cubic (quasi-BCC) nanolattices with the diameter of the nanobeams as small as 34 nm. We show that the compressive yield strengths of quasi-BCC nanolattices even exceed those of their bulk counterparts, despite their relative densities below 0.5. Simultaneously, these quasi-BCC nanolattices exhibit ultrahigh energy absorption capacities, i.e., 100 ± 6 MJ m−3 for gold quasi-BCC nanolattice and 110 ± 10 MJ m−3 for copper quasi-BCC nanolattice. Finite element simulations and theoretical calculations reveal that the deformation of quasi-BCC nanolattice is dominated by nanobeam bending. And the anomalous energy absorption capacities substantially stem from the synergy of the naturally high mechanical strength and plasticity of metals, the size reduction-induced mechanical enhancement, and the quasi-BCC nanolattice architecture. Since the sample size can be scaled up to macroscale at high efficiency and affordable cost, the quasi-BCC nanolattices with ultrahigh energy absorption capacity reported in this work may find great potentials in heat transfer, electric conduction, catalysis applications.},
note = {The fabrication of freestanding 3D lattice structures with beam diameters less than 100 nm is a considerable challenge. Here, the authors report quasi-BCC nanolattices of gold and copper, featuring beam diameters as low as 34 nm, that demonstrate an exceptionally high capacity for energy absorption.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanolattices exhibit attractive mechanical properties such as high strength, high specific strength, and high energy absorption. However, at present, such materials cannot achieve effective fusion of the above properties and scalable production, which hinders their applications in energy conversion and other fields. Herein, we report gold and copper quasi-body centered cubic (quasi-BCC) nanolattices with the diameter of the nanobeams as small as 34 nm. We show that the compressive yield strengths of quasi-BCC nanolattices even exceed those of their bulk counterparts, despite their relative densities below 0.5. Simultaneously, these quasi-BCC nanolattices exhibit ultrahigh energy absorption capacities, i.e., 100 ± 6 MJ m−3 for gold quasi-BCC nanolattice and 110 ± 10 MJ m−3 for copper quasi-BCC nanolattice. Finite element simulations and theoretical calculations reveal that the deformation of quasi-BCC nanolattice is dominated by nanobeam bending. And the anomalous energy absorption capacities substantially stem from the synergy of the naturally high mechanical strength and plasticity of metals, the size reduction-induced mechanical enhancement, and the quasi-BCC nanolattice architecture. Since the sample size can be scaled up to macroscale at high efficiency and affordable cost, the quasi-BCC nanolattices with ultrahigh energy absorption capacity reported in this work may find great potentials in heat transfer, electric conduction, catalysis applications. |
| 47. |  | Rozeline Wijnhorst, Menno Demmenie, Etienne Jambon-Puillet, Freek Ariese, Daniel Bonn, Noushine Shahidzadeh Softness of hydrated salt crystals under deliquescence In: Nature Communications, vol. 14, no. 1090, 2023, (Crystals are commonly associated with their hard and faceted nature. Here, the authors report the transition from hard to soft and deformable, observed in the gradual dissolution of salt crystals that harbor water in their crystalline framework.). @article{nokey,
title = {Softness of hydrated salt crystals under deliquescence},
author = {Rozeline Wijnhorst and Menno Demmenie and Etienne Jambon-Puillet and Freek Ariese and Daniel Bonn and Noushine Shahidzadeh },
url = {https://www.nature.com/articles/s41467-023-36834-0.pdf},
doi = {10.1038/s41467-023-36834-0},
year = {2023},
date = {2023-02-25},
urldate = {2023-02-25},
journal = {Nature Communications},
volume = {14},
number = {1090},
abstract = {Deliquescence is a first-order phase transition, happening when a salt absorbs water vapor. This has a major impact on the stability of crystalline powders that are important for example in pharmacology, food science and for our environment and climate. Here we show that during deliquescence, the abundant salt sodium sulfate decahydrate, mirabilite (Na2SO4·10H2O), behaves differently than anhydrous salts. Using various microscopy techniques combined with Raman spectroscopy, we show that mirabilite crystals not only lose their facets but also become soft and deformable. As a result, microcrystals of mirabilite simultaneously behave crystalline-like in the core bulk and liquid-like at the surface. Defects at the surface can heal at a speed much faster than the deliquescence rate by the mechanism of visco-capillary flow over the surface. While magnesium sulfate hexahydrate (MgSO4⋅6H2O) behaves similarly during deliquescence, a soft and deformable state is completely absent for the anhydrous salts sodium chloride (NaCl) and sodium sulfate thenardite (Na2SO4). The results highlight the effect of crystalline water, and its mobility in the crystalline structure on the observed softness during deliquescence. Controlled hydrated salts have potential applications such as thermal energy storage, where the key parameter is relative humidity rather than temperature.},
note = {Crystals are commonly associated with their hard and faceted nature. Here, the authors report the transition from hard to soft and deformable, observed in the gradual dissolution of salt crystals that harbor water in their crystalline framework.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Deliquescence is a first-order phase transition, happening when a salt absorbs water vapor. This has a major impact on the stability of crystalline powders that are important for example in pharmacology, food science and for our environment and climate. Here we show that during deliquescence, the abundant salt sodium sulfate decahydrate, mirabilite (Na2SO4·10H2O), behaves differently than anhydrous salts. Using various microscopy techniques combined with Raman spectroscopy, we show that mirabilite crystals not only lose their facets but also become soft and deformable. As a result, microcrystals of mirabilite simultaneously behave crystalline-like in the core bulk and liquid-like at the surface. Defects at the surface can heal at a speed much faster than the deliquescence rate by the mechanism of visco-capillary flow over the surface. While magnesium sulfate hexahydrate (MgSO4⋅6H2O) behaves similarly during deliquescence, a soft and deformable state is completely absent for the anhydrous salts sodium chloride (NaCl) and sodium sulfate thenardite (Na2SO4). The results highlight the effect of crystalline water, and its mobility in the crystalline structure on the observed softness during deliquescence. Controlled hydrated salts have potential applications such as thermal energy storage, where the key parameter is relative humidity rather than temperature. |
| 46. |  | Massimo Bocus, Ruben Goeminne, Aran Lamaire, Maarten Cools-Ceuppens, Toon Verstraelen, Veronique Van Speybroeck Nuclear quantum effects on zeolite proton hopping kinetics explored with machine learning potentials and path integral molecular dynamics In: Nature Communications, vol. 14, no. 1008, 2023, (The quantum properties of hydrogen atoms in zeolite-catalyzed reactions are generally neglected due to high computational costs. Here, the authors leverage machine learning to derive accurate quantum kinetics for proton transfer reactions in heterogeneous catalysis.). @article{nokey,
title = {Nuclear quantum effects on zeolite proton hopping kinetics explored with machine learning potentials and path integral molecular dynamics},
author = {Massimo Bocus and Ruben Goeminne and Aran Lamaire and Maarten Cools-Ceuppens and Toon Verstraelen and Veronique Van Speybroeck },
url = {https://www.nature.com/articles/s41467-023-36666-y.pdf},
doi = {10.1038/s41467-023-36666-y},
year = {2023},
date = {2023-02-23},
urldate = {2023-02-23},
journal = {Nature Communications},
volume = {14},
number = {1008},
abstract = {Proton hopping is a key reactive process within zeolite catalysis. However, the accurate determination of its kinetics poses major challenges both for theoreticians and experimentalists. Nuclear quantum effects (NQEs) are known to influence the structure and dynamics of protons, but their rigorous inclusion through the path integral molecular dynamics (PIMD) formalism was so far beyond reach for zeolite catalyzed processes due to the excessive computational cost of evaluating all forces and energies at the Density Functional Theory (DFT) level. Herein, we overcome this limitation by training first a reactive machine learning potential (MLP) that can reproduce with high fidelity the DFT potential energy surface of proton hopping around the first Al coordination sphere in the H-CHA zeolite. The MLP offers an immense computational speedup, enabling us to derive accurate reaction kinetics beyond standard transition state theory for the proton hopping reaction. Overall, more than 0.6 μs of simulation time was needed, which is far beyond reach of any standard DFT approach. NQEs are found to significantly impact the proton hopping kinetics up to ~473 K. Moreover, PIMD simulations with deuterium can be performed without any additional training to compute kinetic isotope effects over a broad range of temperatures.},
note = {The quantum properties of hydrogen atoms in zeolite-catalyzed reactions are generally neglected due to high computational costs. Here, the authors leverage machine learning to derive accurate quantum kinetics for proton transfer reactions in heterogeneous catalysis.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Proton hopping is a key reactive process within zeolite catalysis. However, the accurate determination of its kinetics poses major challenges both for theoreticians and experimentalists. Nuclear quantum effects (NQEs) are known to influence the structure and dynamics of protons, but their rigorous inclusion through the path integral molecular dynamics (PIMD) formalism was so far beyond reach for zeolite catalyzed processes due to the excessive computational cost of evaluating all forces and energies at the Density Functional Theory (DFT) level. Herein, we overcome this limitation by training first a reactive machine learning potential (MLP) that can reproduce with high fidelity the DFT potential energy surface of proton hopping around the first Al coordination sphere in the H-CHA zeolite. The MLP offers an immense computational speedup, enabling us to derive accurate reaction kinetics beyond standard transition state theory for the proton hopping reaction. Overall, more than 0.6 μs of simulation time was needed, which is far beyond reach of any standard DFT approach. NQEs are found to significantly impact the proton hopping kinetics up to ~473 K. Moreover, PIMD simulations with deuterium can be performed without any additional training to compute kinetic isotope effects over a broad range of temperatures. |
| 45. |  | Jacob I. Deneff, Lauren E. S. Rohwer, Kimberly S. Butler, Bryan Kaehr, Dayton J. Vogel, Ting S. Luk, Raphael A. Reyes, Alvaro A. Cruz-Cabrera, James E. Martin, Dorina F. Sava Gallis Orthogonal luminescence lifetime encoding by intermetallic energy transfer in heterometallic rare-earth MOFs In: Nature Communications, vol. 14, no. 981, 2023, (Lifetime-encoded materials are attractive as optical markers. Here, the authors report a design strategy for true orthogonality in encoding based on heterometallic rare-earth MOFs with independently variable lifetime and composition.). @article{nokey,
title = {Orthogonal luminescence lifetime encoding by intermetallic energy transfer in heterometallic rare-earth MOFs},
author = {Jacob I. Deneff and Lauren E. S. Rohwer and Kimberly S. Butler and Bryan Kaehr and Dayton J. Vogel and Ting S. Luk and Raphael A. Reyes and Alvaro A. Cruz-Cabrera and James E. Martin and Dorina F. Sava Gallis },
url = {https://www.nature.com/articles/s41467-023-36576-z.pdf},
doi = {10.1038/s41467-023-36576-z},
year = {2023},
date = {2023-02-22},
urldate = {2023-02-22},
journal = {Nature Communications},
volume = {14},
number = {981},
abstract = {Lifetime-encoded materials are particularly attractive as optical tags, however examples are rare and hindered in practical application by complex interrogation methods. Here, we demonstrate a design strategy towards multiplexed, lifetime-encoded tags via engineering intermetallic energy transfer in a family of heterometallic rare-earth metal-organic frameworks (MOFs). The MOFs are derived from a combination of a high-energy donor (Eu), a low-energy acceptor (Yb) and an optically inactive ion (Gd) with the 1,2,4,5 tetrakis(4-carboxyphenyl) benzene (TCPB) organic linker. Precise manipulation of the luminescence decay dynamics over a wide microsecond regime is achieved via control over metal distribution in these systems. Demonstration of this platform’s relevance as a tag is attained via a dynamic double encoding method that uses the braille alphabet, and by incorporation into photocurable inks patterned on glass and interrogated via digital high-speed imaging. This study reveals true orthogonality in encoding using independently variable lifetime and composition, and highlights the utility of this design strategy, combining facile synthesis and interrogation with complex optical properties.
},
note = {Lifetime-encoded materials are attractive as optical markers. Here, the authors report a design strategy for true orthogonality in encoding based on heterometallic rare-earth MOFs with independently variable lifetime and composition.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Lifetime-encoded materials are particularly attractive as optical tags, however examples are rare and hindered in practical application by complex interrogation methods. Here, we demonstrate a design strategy towards multiplexed, lifetime-encoded tags via engineering intermetallic energy transfer in a family of heterometallic rare-earth metal-organic frameworks (MOFs). The MOFs are derived from a combination of a high-energy donor (Eu), a low-energy acceptor (Yb) and an optically inactive ion (Gd) with the 1,2,4,5 tetrakis(4-carboxyphenyl) benzene (TCPB) organic linker. Precise manipulation of the luminescence decay dynamics over a wide microsecond regime is achieved via control over metal distribution in these systems. Demonstration of this platform’s relevance as a tag is attained via a dynamic double encoding method that uses the braille alphabet, and by incorporation into photocurable inks patterned on glass and interrogated via digital high-speed imaging. This study reveals true orthogonality in encoding using independently variable lifetime and composition, and highlights the utility of this design strategy, combining facile synthesis and interrogation with complex optical properties.
|
| 44. |  | Sabrina D. Eder, Adam Fahy, Matthew G. Barr, J. R. Manson, Bodil Holst, Paul C. Dastoor Sub-resolution contrast in neutral helium microscopy through facet scattering for quantitative imaging of nanoscale topographies on macroscopic surfaces In: Nature Communications, vol. 14, no. 904, 2023, (Neutral helium microscopy is a completely nondestructive, surface-sensitive imaging technique. Here, the authors demonstrate sub-resolution contrast using an advanced facet scattering model to reconstruct the topography of technological thin films in the ångström range.). @article{nokey,
title = {Sub-resolution contrast in neutral helium microscopy through facet scattering for quantitative imaging of nanoscale topographies on macroscopic surfaces},
author = {Sabrina D. Eder and Adam Fahy and Matthew G. Barr and J. R. Manson and Bodil Holst and Paul C. Dastoor},
url = {https://www.nature.com/articles/s41467-023-36578-x.pdf},
doi = {10.1038/s41467-023-36578-x},
year = {2023},
date = {2023-02-17},
urldate = {2023-02-17},
journal = {Nature Communications},
volume = {14},
number = {904},
abstract = {Nanoscale thin film coatings and surface treatments are ubiquitous across industry, science, and engineering; imbuing specific functional or mechanical properties (such as corrosion resistance, lubricity, catalytic activity and electronic behaviour). Non-destructive nanoscale imaging of thin film coatings across large (ca. centimetre) lateral length scales, crucial to a wide range of modern industry, remains a significant technical challenge. By harnessing the unique nature of the helium atom–surface interaction, neutral helium microscopy images these surfaces without altering the sample under investigation. Since the helium atom scatters exclusively from the outermost electronic corrugation of the sample, the technique is completely surface sensitive. Furthermore, with a cross-section that is orders of magnitude larger than that of electrons, neutrons and photons, the probe particle routinely interacts with features down to the scale of surface defects and small adsorbates (including hydrogen). Here, we highlight the capacity of neutral helium microscopy for sub-resolution contrast using an advanced facet scattering model based on nanoscale features. By replicating the observed scattered helium intensities, we demonstrate that sub-resolution contrast arises from the unique surface scattering of the incident probe. Consequently, it is now possible to extract quantitative information from the helium atom image, including localised ångström-scale variations in topography.
},
note = {Neutral helium microscopy is a completely nondestructive, surface-sensitive imaging technique. Here, the authors demonstrate sub-resolution contrast using an advanced facet scattering model to reconstruct the topography of technological thin films in the ångström range.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanoscale thin film coatings and surface treatments are ubiquitous across industry, science, and engineering; imbuing specific functional or mechanical properties (such as corrosion resistance, lubricity, catalytic activity and electronic behaviour). Non-destructive nanoscale imaging of thin film coatings across large (ca. centimetre) lateral length scales, crucial to a wide range of modern industry, remains a significant technical challenge. By harnessing the unique nature of the helium atom–surface interaction, neutral helium microscopy images these surfaces without altering the sample under investigation. Since the helium atom scatters exclusively from the outermost electronic corrugation of the sample, the technique is completely surface sensitive. Furthermore, with a cross-section that is orders of magnitude larger than that of electrons, neutrons and photons, the probe particle routinely interacts with features down to the scale of surface defects and small adsorbates (including hydrogen). Here, we highlight the capacity of neutral helium microscopy for sub-resolution contrast using an advanced facet scattering model based on nanoscale features. By replicating the observed scattered helium intensities, we demonstrate that sub-resolution contrast arises from the unique surface scattering of the incident probe. Consequently, it is now possible to extract quantitative information from the helium atom image, including localised ångström-scale variations in topography.
|
| 43. |  | Yi Yang, Jong Bin Kim, Seong Kyeong Nam, Mengmeng Zhang, Jiangping Xu, Jintao Zhu, Shin-Hyun Kim Nanostructure-free crescent-shaped microparticles as full-color reflective pigments In: Nature Communications, vol. 14, no. 793, 2023, (Structural colors arise from nanostructures or special microgeometries. Here, the authors report on crescent-shaped microparticles as full-color reflective pigments that show brilliant colors in directional light and are transparent in ambient light.). @article{nokey,
title = {Nanostructure-free crescent-shaped microparticles as full-color reflective pigments},
author = {Yi Yang and Jong Bin Kim and Seong Kyeong Nam and Mengmeng Zhang and Jiangping Xu and Jintao Zhu and Shin-Hyun Kim },
url = {https://www.nature.com/articles/s41467-023-36482-4.pdf},
doi = {10.1038/s41467-023-36482-4},
year = {2023},
date = {2023-02-11},
urldate = {2023-02-11},
journal = {Nature Communications},
volume = {14},
number = {793},
abstract = {Structural colors provide a promising visualization with high color saturation, iridescent characteristics, and fade resistance. However, pragmatic uses are frequently impeded by complex manufacturing processes for sophisticated nanostructures. Here, we report a facile emulsion-templating strategy to produce crescent-shaped microparticles as structural color pigments. The micro-crescents exhibit brilliant colors under directional light originating from total internal reflections and optical interferences in the absence of periodic nanostructures while being transparent under ambient light. The colors are finely tunable by adjusting the size of the micro-crescents, which can be further mixed to enrich the variety. Importantly, the pre-defined convex surface secures high stability of colors and enables structural coloration on target surfaces through direct deposition as inks. We anticipate this class of nanostructure-free structural colorants is pragmatic as invisible inks in particular for anti-counterfeiting patches and color cosmetics with distinctive impressions due to low-cost, scalable manufacturing, unique optical properties, and versatility.},
note = {Structural colors arise from nanostructures or special microgeometries. Here, the authors report on crescent-shaped microparticles as full-color reflective pigments that show brilliant colors in directional light and are transparent in ambient light.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Structural colors provide a promising visualization with high color saturation, iridescent characteristics, and fade resistance. However, pragmatic uses are frequently impeded by complex manufacturing processes for sophisticated nanostructures. Here, we report a facile emulsion-templating strategy to produce crescent-shaped microparticles as structural color pigments. The micro-crescents exhibit brilliant colors under directional light originating from total internal reflections and optical interferences in the absence of periodic nanostructures while being transparent under ambient light. The colors are finely tunable by adjusting the size of the micro-crescents, which can be further mixed to enrich the variety. Importantly, the pre-defined convex surface secures high stability of colors and enables structural coloration on target surfaces through direct deposition as inks. We anticipate this class of nanostructure-free structural colorants is pragmatic as invisible inks in particular for anti-counterfeiting patches and color cosmetics with distinctive impressions due to low-cost, scalable manufacturing, unique optical properties, and versatility. |
| 42. |  | Yuan Zhong, Jiangwei Zhang, Tingting Li, Wenwu Xu, Qiaofeng Yao, Min Lu, Xue Bai, Zhennan Wu, Jianping Xie, Yu Zhang Suppression of kernel vibrations by layer-by-layer ligand engineering boosts photoluminescence efficiency of gold nanoclusters In: Nature Communications, vol. 14, no. 658, 2023, (The photoluminescence of gold nanoclusters is affected by low-frequency acoustic vibrations. Here, the authors demonstrate that layer-by-layer ligand engineering can suppress such structural vibrations to achieve brighter emissions.). @article{nokey,
title = {Suppression of kernel vibrations by layer-by-layer ligand engineering boosts photoluminescence efficiency of gold nanoclusters},
author = {Yuan Zhong and Jiangwei Zhang and Tingting Li and Wenwu Xu and Qiaofeng Yao and Min Lu and Xue Bai and Zhennan Wu and Jianping Xie and Yu Zhang },
url = {https://www.nature.com/articles/s41467-023-36387-2.pdf},
doi = {10.1038/s41467-023-36387-2},
year = {2023},
date = {2023-02-07},
urldate = {2023-02-07},
journal = {Nature Communications},
volume = {14},
number = {658},
abstract = {The restriction of structural vibration has assumed great importance in attaining bright emission of luminescent metal nanoclusters (NCs), where tremendous efforts are devoted to manipulating the surface landscape yet remain challenges for modulation of the structural vibration of the metal kernel. Here, we report efficient suppression of kernel vibration achieving enhancement in emission intensity, by rigidifying the surface of metal NCs and propagating as-developed strains into the metal core. Specifically, a layer-by-layer triple-ligands surface engineering is deployed to allow the solution-phase Au NCs with strong metal core-dictated fluorescence, up to the high absolute quantum yields of 90.3 ± 3.5%. The as-rigidified surface imposed by synergistic supramolecular interactions greatly influences the low-frequency acoustic vibration of the metal kernel, resulting in a subtle change in vibration frequency but a reduction in amplitude of oscillation. This scenario therewith impedes the non-radiative relaxation of electron dynamics, rendering the Au NCs with strong emission. The presented study exemplifies the linkage between surface chemistry and core-state emission of metal NCs, and proposes a strategy for brighter emitting metal NCs by regulating their interior metal core-involved motion.},
note = {The photoluminescence of gold nanoclusters is affected by low-frequency acoustic vibrations. Here, the authors demonstrate that layer-by-layer ligand engineering can suppress such structural vibrations to achieve brighter emissions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The restriction of structural vibration has assumed great importance in attaining bright emission of luminescent metal nanoclusters (NCs), where tremendous efforts are devoted to manipulating the surface landscape yet remain challenges for modulation of the structural vibration of the metal kernel. Here, we report efficient suppression of kernel vibration achieving enhancement in emission intensity, by rigidifying the surface of metal NCs and propagating as-developed strains into the metal core. Specifically, a layer-by-layer triple-ligands surface engineering is deployed to allow the solution-phase Au NCs with strong metal core-dictated fluorescence, up to the high absolute quantum yields of 90.3 ± 3.5%. The as-rigidified surface imposed by synergistic supramolecular interactions greatly influences the low-frequency acoustic vibration of the metal kernel, resulting in a subtle change in vibration frequency but a reduction in amplitude of oscillation. This scenario therewith impedes the non-radiative relaxation of electron dynamics, rendering the Au NCs with strong emission. The presented study exemplifies the linkage between surface chemistry and core-state emission of metal NCs, and proposes a strategy for brighter emitting metal NCs by regulating their interior metal core-involved motion. |
| 41. |  | Yong Chen, Satoshi Takeya, Amadeu K. Sum Topological dual and extended relations between networks of clathrate hydrates and Frank-Kasper phases In: Nature Communications, vol. 14, no. 596, 2023, (Topological dual and extended relations between networks of clathrate hydrates and Frank-Kasper phases Clathrate hydrates are topological duals of Frank-Kasper phases. Here, the authors employ MD simulations to provide an alternative way to explore the intrinsic structural relationships of H-bonded networks of clathrate hydrates and other unrelated ordered materials.). @article{nokey,
title = {Topological dual and extended relations between networks of clathrate hydrates and Frank-Kasper phases},
author = {Yong Chen and Satoshi Takeya and Amadeu K. Sum },
url = {https://www.nature.com/articles/s41467-023-36242-4.pdf},
doi = {10.1038/s41467-023-36242-4},
year = {2023},
date = {2023-02-03},
urldate = {2023-02-03},
journal = {Nature Communications},
volume = {14},
number = {596},
abstract = {Clathrate hydrates are a class of ordered structures that are stabilized via the delicate balance of hydrophobic interactions between water and guest molecules, of which the space-filling network of hydrogen-bonded (H-bonded) water molecules are closely related to tetrahedrally close-packed structures, known as Frank-Kasper (FK) phases. Here we report an alternative way to understand the intricate structures of clathrate hydrates, which unveils the diverse crystalline H-bonded networks that can be generated via assembly of one common building block. In addition to the intrinsic relations and pathways linking these crystals, we further illustrate the rich structural possibilities of clathrate hydrates. Given that the topological dual relations between networks of clathrate hydrates and tetrahedral close-packed structures, the descriptors presented for clathrate hydrates can be directly extended to other ordered materials for a more thorough understanding of their nucleation, phases transition, and co-existence mechanisms.},
note = {Topological dual and extended relations between networks of clathrate hydrates and Frank-Kasper phases Clathrate hydrates are topological duals of Frank-Kasper phases. Here, the authors employ MD simulations to provide an alternative way to explore the intrinsic structural relationships of H-bonded networks of clathrate hydrates and other unrelated ordered materials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Clathrate hydrates are a class of ordered structures that are stabilized via the delicate balance of hydrophobic interactions between water and guest molecules, of which the space-filling network of hydrogen-bonded (H-bonded) water molecules are closely related to tetrahedrally close-packed structures, known as Frank-Kasper (FK) phases. Here we report an alternative way to understand the intricate structures of clathrate hydrates, which unveils the diverse crystalline H-bonded networks that can be generated via assembly of one common building block. In addition to the intrinsic relations and pathways linking these crystals, we further illustrate the rich structural possibilities of clathrate hydrates. Given that the topological dual relations between networks of clathrate hydrates and tetrahedral close-packed structures, the descriptors presented for clathrate hydrates can be directly extended to other ordered materials for a more thorough understanding of their nucleation, phases transition, and co-existence mechanisms. |
| 40. |  | Ahmed A. Agiza, Kady Oakley, Jacob K. Rosenstein, Brenda M. Rubenstein, Eunsuk Kim, Marc Riedel, Sherief Reda Digital circuits and neural networks based on acid-base chemistry implemented by robotic fluid handling In: Nature Communications, vol. 14, no. 496, 2023, (The complementarity of acids and bases is a fundamental chemical concept. Here, the authors use simple acid-base chemistry to encode binary information and perform information processing including digital circuits and neural networks using robotic fluid handling.). @article{nokey,
title = {Digital circuits and neural networks based on acid-base chemistry implemented by robotic fluid handling},
author = {Ahmed A. Agiza and Kady Oakley and Jacob K. Rosenstein and Brenda M. Rubenstein and Eunsuk Kim and Marc Riedel and Sherief Reda },
url = {https://www.nature.com/articles/s41467-023-36206-8.pdf},
doi = {10.1038/s41467-023-36206-8},
year = {2023},
date = {2023-01-30},
urldate = {2023-01-30},
journal = {Nature Communications},
volume = {14},
number = {496},
abstract = {Acid-base reactions are ubiquitous, easy to prepare, and execute without sophisticated equipment. Acids and bases are also inherently complementary and naturally map to a universal representation of “0” and “1.” Here, we propose how to leverage acids, bases, and their reactions to encode binary information and perform information processing based upon the majority and negation operations. These operations form a functionally complete set that we use to implement more complex computations such as digital circuits and neural networks. We present the building blocks needed to build complete digital circuits using acids and bases for dual-rail encoding data values as complementary pairs, including a set of primitive logic functions that are widely applicable to molecular computation. We demonstrate how to implement neural network classifiers and some classes of digital circuits with acid-base reactions orchestrated by a robotic fluid handling device. We validate the neural network experimentally on a number of images with different formats, resulting in a perfect match to the in-silico classifier. Additionally, the simulation of our acid-base classifier matches the results of the in-silico classifier with approximately 99% similarity.},
note = {The complementarity of acids and bases is a fundamental chemical concept. Here, the authors use simple acid-base chemistry to encode binary information and perform information processing including digital circuits and neural networks using robotic fluid handling.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Acid-base reactions are ubiquitous, easy to prepare, and execute without sophisticated equipment. Acids and bases are also inherently complementary and naturally map to a universal representation of “0” and “1.” Here, we propose how to leverage acids, bases, and their reactions to encode binary information and perform information processing based upon the majority and negation operations. These operations form a functionally complete set that we use to implement more complex computations such as digital circuits and neural networks. We present the building blocks needed to build complete digital circuits using acids and bases for dual-rail encoding data values as complementary pairs, including a set of primitive logic functions that are widely applicable to molecular computation. We demonstrate how to implement neural network classifiers and some classes of digital circuits with acid-base reactions orchestrated by a robotic fluid handling device. We validate the neural network experimentally on a number of images with different formats, resulting in a perfect match to the in-silico classifier. Additionally, the simulation of our acid-base classifier matches the results of the in-silico classifier with approximately 99% similarity. |
| 39. | 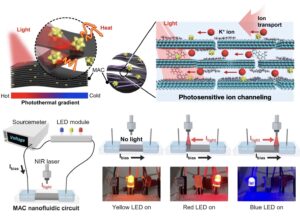 | Jeonghee Yeom, Ayoung Choe, Jiyun Lee, Jeeyoon Kim, Jinyoung Kimand Seung Hak Ohand Cheolhong Park, Sangyun Naand Young-Eun Shinand Youngoh Lee, Yun Goo Ro, Sang Kyu Kwak, Hyunhyub Ko Photosensitive ion channels in layered MXene membranes modified with plasmonic gold nanostars and cellulose nanofibers In: Nature Communications, vol. 14, no. 359, 2023, (Artificial ion channels are in demand for ionotronic devices. Here, the authors use layered MXene membranes modified with plasmonic gold nanostars and cellulose nanofibers to convert a thermal gradient into an ion current for photosensitive ion channeling.). @article{nokey,
title = {Photosensitive ion channels in layered MXene membranes modified with plasmonic gold nanostars and cellulose nanofibers},
author = {Jeonghee Yeom and Ayoung Choe and Jiyun Lee and Jeeyoon Kim and Jinyoung Kimand Seung Hak Ohand Cheolhong Park and Sangyun Naand Young-Eun Shinand Youngoh Lee and Yun Goo Ro and Sang Kyu Kwak and Hyunhyub Ko},
url = {https://www.nature.com/articles/s41467-023-36039-5.pdf},
doi = {10.1038/s41467-023-36039-5},
year = {2023},
date = {2023-01-23},
urldate = {2023-01-23},
journal = {Nature Communications},
volume = {14},
number = {359},
abstract = {Ion channels transduce external stimuli into ion-transport-mediated signaling, which has received considerable attention in diverse fields such as sensors, energy harvesting devices, and desalination membrane. In this work, we present a photosensitive ion channel based on plasmonic gold nanostars (AuNSs) and cellulose nanofibers (CNFs) embedded in layered MXene nanosheets. The MXene/AuNS/CNF (MAC) membrane provides subnanometer-sized ionic pathways for light-sensitive cationic flow. When the MAC nanochannel is exposed to NIR light, a photothermal gradient is formed, which induces directional photothermo-osmotic flow of nanoconfined electrolyte against the thermal gradient and produces a net ionic current. MAC membrane exhibits enhanced photothermal current compared with pristine MXene, which is attributed to the combined photothermal effects of plasmonic AuNSs and MXene and the widened interspacing of the MAC composite via the hydrophilic nanofibrils. The MAC composite membranes are envisioned to be applied in flexible ionic channels with ionogels and light-controlled ionic circuits.},
note = {Artificial ion channels are in demand for ionotronic devices. Here, the authors use layered MXene membranes modified with plasmonic gold nanostars and cellulose nanofibers to convert a thermal gradient into an ion current for photosensitive ion channeling.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ion channels transduce external stimuli into ion-transport-mediated signaling, which has received considerable attention in diverse fields such as sensors, energy harvesting devices, and desalination membrane. In this work, we present a photosensitive ion channel based on plasmonic gold nanostars (AuNSs) and cellulose nanofibers (CNFs) embedded in layered MXene nanosheets. The MXene/AuNS/CNF (MAC) membrane provides subnanometer-sized ionic pathways for light-sensitive cationic flow. When the MAC nanochannel is exposed to NIR light, a photothermal gradient is formed, which induces directional photothermo-osmotic flow of nanoconfined electrolyte against the thermal gradient and produces a net ionic current. MAC membrane exhibits enhanced photothermal current compared with pristine MXene, which is attributed to the combined photothermal effects of plasmonic AuNSs and MXene and the widened interspacing of the MAC composite via the hydrophilic nanofibrils. The MAC composite membranes are envisioned to be applied in flexible ionic channels with ionogels and light-controlled ionic circuits. |
| 38. | 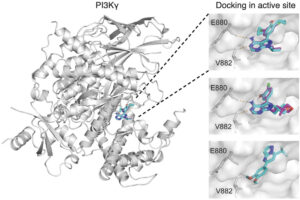 | Michael Moret, Irene Pachon Angona, Leandro Cotos, Shen Yan, Kenneth Atz, Cyrill Brunner, Martin Baumgartner, Francesca Grisoni, Gisbert Schneider Leveraging molecular structure and bioactivity with chemical language models for de novo drug design In: Nature Communications, vol. 14, no. 114, 2023, (Generative Deep Learning holds promise for mining the unexplored "chemical universe" for new drugs. Here, the authors demonstrate the de novo design of phosphoinositide 3-kinase gamma (PI3Kγ) inhibitors for the PI3K/Akt pathway in human tumor cells.). @article{nokey,
title = {Leveraging molecular structure and bioactivity with chemical language models for de novo drug design},
author = {Michael Moret and Irene Pachon Angona and Leandro Cotos and Shen Yan and Kenneth Atz and Cyrill Brunner and Martin Baumgartner and Francesca Grisoni and Gisbert Schneider },
url = {https://www.nature.com/articles/s41467-022-35692-6.pdf},
doi = {10.1038/s41467-022-35692-6},
year = {2023},
date = {2023-01-07},
urldate = {2023-01-07},
journal = {Nature Communications},
volume = {14},
number = {114},
abstract = {Generative chemical language models (CLMs) can be used for de novo molecular structure generation by learning from a textual representation of molecules. Here, we show that hybrid CLMs can additionally leverage the bioactivity information available for the training compounds. To computationally design ligands of phosphoinositide 3-kinase gamma (PI3Kγ), a collection of virtual molecules was created with a generative CLM. This virtual compound library was refined using a CLM-based classifier for bioactivity prediction. This second hybrid CLM was pretrained with patented molecular structures and fine-tuned with known PI3Kγ ligands. Several of the computer-generated molecular designs were commercially available, enabling fast prescreening and preliminary experimental validation. A new PI3Kγ ligand with sub-micromolar activity was identified, highlighting the method’s scaffold-hopping potential. Chemical synthesis and biochemical testing of two of the top-ranked de novo designed molecules and their derivatives corroborated the model’s ability to generate PI3Kγ ligands with medium to low nanomolar activity for hit-to-lead expansion. The most potent compounds led to pronounced inhibition of PI3K-dependent Akt phosphorylation in a medulloblastoma cell model, demonstrating efficacy of PI3Kγ ligands in PI3K/Akt pathway repression in human tumor cells. The results positively advocate hybrid CLMs for virtual compound screening and activity-focused molecular design.},
note = {Generative Deep Learning holds promise for mining the unexplored "chemical universe" for new drugs. Here, the authors demonstrate the de novo design of phosphoinositide 3-kinase gamma (PI3Kγ) inhibitors for the PI3K/Akt pathway in human tumor cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Generative chemical language models (CLMs) can be used for de novo molecular structure generation by learning from a textual representation of molecules. Here, we show that hybrid CLMs can additionally leverage the bioactivity information available for the training compounds. To computationally design ligands of phosphoinositide 3-kinase gamma (PI3Kγ), a collection of virtual molecules was created with a generative CLM. This virtual compound library was refined using a CLM-based classifier for bioactivity prediction. This second hybrid CLM was pretrained with patented molecular structures and fine-tuned with known PI3Kγ ligands. Several of the computer-generated molecular designs were commercially available, enabling fast prescreening and preliminary experimental validation. A new PI3Kγ ligand with sub-micromolar activity was identified, highlighting the method’s scaffold-hopping potential. Chemical synthesis and biochemical testing of two of the top-ranked de novo designed molecules and their derivatives corroborated the model’s ability to generate PI3Kγ ligands with medium to low nanomolar activity for hit-to-lead expansion. The most potent compounds led to pronounced inhibition of PI3K-dependent Akt phosphorylation in a medulloblastoma cell model, demonstrating efficacy of PI3Kγ ligands in PI3K/Akt pathway repression in human tumor cells. The results positively advocate hybrid CLMs for virtual compound screening and activity-focused molecular design. |
| 37. |  | Yingying Jiang, Martial Duchamp, Shi Jun Ang, Hongwei Yan, Teck Leong Tan, Utkur Mirsaidov Dynamics of the fcc-to-bcc phase transition in single-crystalline PdCu alloy nanoparticles In: Nature Communications, vol. 14, no. 104, 2023, (Phase transitions in crystals are challenging to study with atomic resolution. Here, the authors reveal that the transition from fcc to bcc occurs across a coherent interface only a few atoms wide, which serves as a precursor phase for the nucleation of the bcc phase.). @article{nokey,
title = {Dynamics of the fcc-to-bcc phase transition in single-crystalline PdCu alloy nanoparticles},
author = {Yingying Jiang and Martial Duchamp and Shi Jun Ang and Hongwei Yan and Teck Leong Tan and Utkur Mirsaidov },
url = {https://www.nature.com/articles/s41467-022-35325-y.pdf},
doi = {10.1038/s41467-022-35325-y},
year = {2023},
date = {2023-01-06},
urldate = {2023-01-06},
journal = {Nature Communications},
volume = {14},
number = {104},
abstract = {Two most common crystal structures in metals and metal alloys are body-centered cubic (bcc) and face-centered cubic (fcc) structures. The phase transitions between these structures play an important role in the production of durable and functional metal alloys. Despite their technological significance, the details of such phase transitions are largely unknown because of the challenges associated with probing these processes. Here, we describe the nanoscopic details of an fcc-to-bcc phase transition in PdCu alloy nanoparticles (NPs) using in situ heating transmission electron microscopy. Our observations reveal that the bcc phase always nucleates from the edge of the fcc NP, and then propagates across the NP by forming a distinct few-atoms-wide coherent bcc–fcc interface. Notably, this interface acts as an intermediate precursor phase for the nucleation of a bcc phase. These insights into the fcc-to-bcc phase transition are important for understanding solid − solid phase transitions in general and can help to tailor the functional properties of metals and their alloys.},
note = {Phase transitions in crystals are challenging to study with atomic resolution. Here, the authors reveal that the transition from fcc to bcc occurs across a coherent interface only a few atoms wide, which serves as a precursor phase for the nucleation of the bcc phase.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Two most common crystal structures in metals and metal alloys are body-centered cubic (bcc) and face-centered cubic (fcc) structures. The phase transitions between these structures play an important role in the production of durable and functional metal alloys. Despite their technological significance, the details of such phase transitions are largely unknown because of the challenges associated with probing these processes. Here, we describe the nanoscopic details of an fcc-to-bcc phase transition in PdCu alloy nanoparticles (NPs) using in situ heating transmission electron microscopy. Our observations reveal that the bcc phase always nucleates from the edge of the fcc NP, and then propagates across the NP by forming a distinct few-atoms-wide coherent bcc–fcc interface. Notably, this interface acts as an intermediate precursor phase for the nucleation of a bcc phase. These insights into the fcc-to-bcc phase transition are important for understanding solid − solid phase transitions in general and can help to tailor the functional properties of metals and their alloys. |
| 36. | 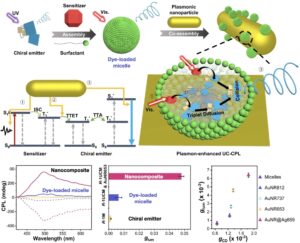 | Tonghan Zhao, Dejing Meng, Zhijian Hu, Wenjing Sun, Yinglu Ji, Jianlei Han, Xue Jin, Xiaochun Wu, Pengfei Duan Enhanced chiroptic properties of nanocomposites of achiral plasmonic nanoparticles decorated with chiral dye-loaded micelles In: Nature Communications, vol. 14, no. 81, 2023, (Circularly polarized luminescent materials with large dissymmetry and efficiency are in demand. Here, the authors investigate chirality-induced spin polarization in achiral gold nanorods decorated with chiral dye-loaded micelles to enhance chiroptic activity.). @article{nokey,
title = {Enhanced chiroptic properties of nanocomposites of achiral plasmonic nanoparticles decorated with chiral dye-loaded micelles},
author = {Tonghan Zhao and Dejing Meng and Zhijian Hu and Wenjing Sun and Yinglu Ji and Jianlei Han and Xue Jin and Xiaochun Wu and Pengfei Duan},
url = {https://www.nature.com/articles/s41467-022-35699-z.pdf},
doi = {10.1038/s41467-022-35699-z},
year = {2023},
date = {2023-01-05},
urldate = {2023-01-05},
journal = {Nature Communications},
volume = {14},
number = {81},
abstract = {The development of circularly polarized luminescence (CPL)-active materials with both large luminescence dissymmetry factor (glum) and high emission efficiency continues to be a major challenge. Here, we present an approach to improve the overall CPL performance by integrating triplet-triplet annihila- tion-based photon upconversion (TTA-UC) with localized surface plasmon resonance. Dye-loaded chiral micelles possessing TTA-UC ability are designed and attached on the surface of achiral gold nanorods (AuNRs). The long- itudinal and transversal resonance peaks of AuNRs overlap with the absorption and emission of dye-loaded chiral micelles, respectively. Typically, 43-fold amplification of glum value accompanied by 3-fold enhancement of upcon- version are obtained simultaneously when Au@Ag nanorods are employed in the composites. More importantly, transient absorption spectra reveal a fast accumulation of spin-polarized triplet excitons in the composites. Therefore, the enhancement of chirality-induced spin polarization should be in charge of the amplification of glum value. Our design strategy suggests that combining plasmonic nanomaterials with chiral organic materials could aid in the devel- opment of chiroptical nanomaterials.},
note = {Circularly polarized luminescent materials with large dissymmetry and efficiency are in demand. Here, the authors investigate chirality-induced spin polarization in achiral gold nanorods decorated with chiral dye-loaded micelles to enhance chiroptic activity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The development of circularly polarized luminescence (CPL)-active materials with both large luminescence dissymmetry factor (glum) and high emission efficiency continues to be a major challenge. Here, we present an approach to improve the overall CPL performance by integrating triplet-triplet annihila- tion-based photon upconversion (TTA-UC) with localized surface plasmon resonance. Dye-loaded chiral micelles possessing TTA-UC ability are designed and attached on the surface of achiral gold nanorods (AuNRs). The long- itudinal and transversal resonance peaks of AuNRs overlap with the absorption and emission of dye-loaded chiral micelles, respectively. Typically, 43-fold amplification of glum value accompanied by 3-fold enhancement of upcon- version are obtained simultaneously when Au@Ag nanorods are employed in the composites. More importantly, transient absorption spectra reveal a fast accumulation of spin-polarized triplet excitons in the composites. Therefore, the enhancement of chirality-induced spin polarization should be in charge of the amplification of glum value. Our design strategy suggests that combining plasmonic nanomaterials with chiral organic materials could aid in the devel- opment of chiroptical nanomaterials. |
| 35. | 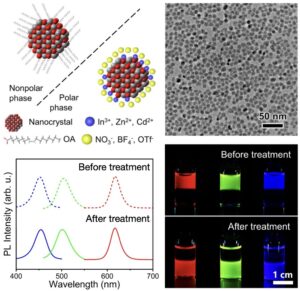 | Pengwei Xiao, Zhoufan Zhang, Junjun Ge, Yalei Deng, Xufeng Chen, Jian-Rong Zhang, Zhengtao Deng, Yu Kambe, Dmitri V. Talapin, Yuanyuan Wang Surface passivation of intensely luminescent all-inorganic nanocrystals and their direct optical patterning In: Nature Communications, vol. 14, no. 49, 2023, (All-inorganic nanocrystals are of great importance for a variety of electronic applications. Here, the authors use metal salts to remove organic ligands to obtain passivated nanocrystals with improved fluorescence yield for direct optical patterning.). @article{nokey,
title = {Surface passivation of intensely luminescent all-inorganic nanocrystals and their direct optical patterning},
author = {Pengwei Xiao and Zhoufan Zhang and Junjun Ge and Yalei Deng and Xufeng Chen and Jian-Rong Zhang and Zhengtao Deng and Yu Kambe and Dmitri V. Talapin and Yuanyuan Wang },
url = {https://www.nature.com/articles/s41467-022-35702-7.pdf},
doi = {10.1038/s41467-022-35702-7},
year = {2023},
date = {2023-01-04},
urldate = {2023-01-04},
journal = {Nature Communications},
volume = {14},
number = {49},
abstract = {All-inorganic nanocrystals (NCs) are of great importance in a range of electronic devices. However, current all-inorganic NCs suffer from limitations in their optical properties, such as low fluorescence efficiencies. Here, we develop a general surface treatment strategy to obtain intensely luminescent all-inorganic NCs (ILANs) by using designed metal salts with noncoordinating anions that play a dual role in the surface treatment process: (i) removing the original organic ligands and (ii) binding to unpassivated Lewis basic sites to preserve the photoluminescent (PL) properties of the NCs. The absolute photoluminescence quantum yields (PLQYs) of red-emitting CdSe/ZnS NCs, green-emitting CdSe/CdZnSeS/ZnS NCs and blue-emitting CdZnS/ZnS NCs in polar solvents are 97%, 80% and 72%, respectively. Further study reveals that the passivated Lewis basic sites of ILANs by metal cations boost the efficiency of radiative recombination of electron-hole pairs. While the passivation of Lewis basic sites leads to a high PLQY of ILANs, the exposed Lewis acidic sites provide the possibility for in situ tuning of the functions of NCs, creating opportunities for direct optical patterning of functional NCs with high resolution.},
note = {All-inorganic nanocrystals are of great importance for a variety of electronic applications. Here, the authors use metal salts to remove organic ligands to obtain passivated nanocrystals with improved fluorescence yield for direct optical patterning.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
All-inorganic nanocrystals (NCs) are of great importance in a range of electronic devices. However, current all-inorganic NCs suffer from limitations in their optical properties, such as low fluorescence efficiencies. Here, we develop a general surface treatment strategy to obtain intensely luminescent all-inorganic NCs (ILANs) by using designed metal salts with noncoordinating anions that play a dual role in the surface treatment process: (i) removing the original organic ligands and (ii) binding to unpassivated Lewis basic sites to preserve the photoluminescent (PL) properties of the NCs. The absolute photoluminescence quantum yields (PLQYs) of red-emitting CdSe/ZnS NCs, green-emitting CdSe/CdZnSeS/ZnS NCs and blue-emitting CdZnS/ZnS NCs in polar solvents are 97%, 80% and 72%, respectively. Further study reveals that the passivated Lewis basic sites of ILANs by metal cations boost the efficiency of radiative recombination of electron-hole pairs. While the passivation of Lewis basic sites leads to a high PLQY of ILANs, the exposed Lewis acidic sites provide the possibility for in situ tuning of the functions of NCs, creating opportunities for direct optical patterning of functional NCs with high resolution. |
| 34. | 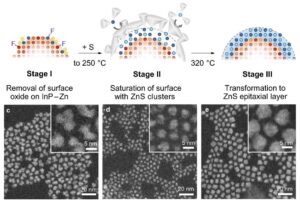 | Yeongho Choi, Donghyo Hahm, Wan Ki Bae, Jaehoon Lim Heteroepitaxial chemistry of zinc chalcogenides on InP nanocrystals for defect-free interfaces with atomic uniformity In: Nature Communications, vol. 14, no. 43, 2023, (Heteroepitaxy on colloidal nanocrystals often yields defective heterostructures due to intricate reaction pathways. Here, the authors decode the surface chemistry at the molecular level to realize defect-free interfaces with atomic uniformity.). @article{nokey,
title = {Heteroepitaxial chemistry of zinc chalcogenides on InP nanocrystals for defect-free interfaces with atomic uniformity},
author = {Yeongho Choi and Donghyo Hahm and Wan Ki Bae and Jaehoon Lim },
url = {https://www.nature.com/articles/s41467-022-35731-2.pdf},
doi = {10.1038/s41467-022-35731-2},
year = {2023},
date = {2023-01-03},
urldate = {2023-01-03},
journal = {Nature Communications},
volume = {14},
number = {43},
abstract = {Heteroepitaxy on colloidal semiconductor nanocrystals is an essential strategy for manipulating their optoelectronic functionalities. However, their practical synthesis typically leads to scattered and unexpected outcomes due to the intervention of multiple reaction pathways associated with complicated side products of reactants. Here, the heteroepitaxy mechanism of zinc chalcogenide initiated on indium phosphide (InP) colloidal nanocrystals is elucidated using the precursors, zinc carboxylate and trialkylphosphine selenide. The high magnetic receptivity of 77Se and the characteristic longitudinal optical phonon mode of ZnSe allowed for monitoring the sequence of epilayer formation at the molecular level. The investigation revealed the sterically hindered acyloxytrialkylphosphonium and diacyloxytrialkylphosphorane to be main intermediates in the surface reaction, which retards the metal ion adsorption by a large steric hindrance. The transformation of adsorbates to the crystalline epilayer was disturbed by surface oxides. Raman scattering disclosed the pathway of secondary surface oxidation triggered by carboxylate ligands migrated from zinc carboxylate. The surface-initiated heteroepitaxy protocol is proposed to fabricate core/shell heterostructured nanocrystals with atomic-scale uniformity of epilayers. Despite the large lattice mismatch of ZnS to InP, we realised a uniform and interface defect-free ZnS epilayer (~0.3 nm thickness) on InP nanocrystals, as evidenced by a high photoluminescence quantum yield of 97.3%.},
note = {Heteroepitaxy on colloidal nanocrystals often yields defective heterostructures due to intricate reaction pathways. Here, the authors decode the surface chemistry at the molecular level to realize defect-free interfaces with atomic uniformity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Heteroepitaxy on colloidal semiconductor nanocrystals is an essential strategy for manipulating their optoelectronic functionalities. However, their practical synthesis typically leads to scattered and unexpected outcomes due to the intervention of multiple reaction pathways associated with complicated side products of reactants. Here, the heteroepitaxy mechanism of zinc chalcogenide initiated on indium phosphide (InP) colloidal nanocrystals is elucidated using the precursors, zinc carboxylate and trialkylphosphine selenide. The high magnetic receptivity of 77Se and the characteristic longitudinal optical phonon mode of ZnSe allowed for monitoring the sequence of epilayer formation at the molecular level. The investigation revealed the sterically hindered acyloxytrialkylphosphonium and diacyloxytrialkylphosphorane to be main intermediates in the surface reaction, which retards the metal ion adsorption by a large steric hindrance. The transformation of adsorbates to the crystalline epilayer was disturbed by surface oxides. Raman scattering disclosed the pathway of secondary surface oxidation triggered by carboxylate ligands migrated from zinc carboxylate. The surface-initiated heteroepitaxy protocol is proposed to fabricate core/shell heterostructured nanocrystals with atomic-scale uniformity of epilayers. Despite the large lattice mismatch of ZnS to InP, we realised a uniform and interface defect-free ZnS epilayer (~0.3 nm thickness) on InP nanocrystals, as evidenced by a high photoluminescence quantum yield of 97.3%. |
| 33. |  | Yilong Zhou, Gaurav Arya
Discovery of two-dimensional binary nanoparticle superlattices using global Monte Carlo optimization In: Nature Communications, vol. 13, no. 7976, 2022, (Binary nanoparticle superlattices exhibit different collective optical, magnetic, and electronic properties. Here, the authors develop an efficient global optimization algorithm for the discovery of periodic 2D architectures forming at fluid interfaces.). @article{nokey,
title = {Discovery of two-dimensional binary nanoparticle superlattices using global Monte Carlo optimization},
author = {Yilong Zhou and Gaurav Arya
},
url = {https://www.nature.com/articles/s41467-022-35690-8.pdf},
doi = {10.1038/s41467-022-35690-8},
year = {2022},
date = {2022-12-29},
urldate = {2022-12-29},
journal = {Nature Communications},
volume = {13},
number = {7976},
abstract = {Binary nanoparticle (NP) superlattices exhibit distinct collective plasmonic, magnetic, optical, and electronic properties. Here, we computationally demonstrate how fluid-fluid interfaces could be used to self-assemble binary systems of NPs into 2D superlattices when the NP species exhibit different miscibility with the fluids forming the interface. We develop a basin-hopping Monte Carlo (BHMC) algorithm tailored for interface-trapped structures to rapidly determine the ground-state configuration of NPs, allowing us to explore the repertoire of binary NP architectures formed at the interface. By varying the NP size ratio, interparticle interaction strength, and difference in NP miscibility with the two fluids, we demonstrate the assembly of an array of exquisite 2D periodic architectures, including AB-, AB2-, and AB3-type monolayer superlattices as well as AB-, AB2-, A3B5-, and A4B6-type bilayer superlattices. Our results suggest that the interfacial assembly approach could be a versatile platform for fabricating 2D colloidal superlattices with tunable structure and properties.},
note = {Binary nanoparticle superlattices exhibit different collective optical, magnetic, and electronic properties. Here, the authors develop an efficient global optimization algorithm for the discovery of periodic 2D architectures forming at fluid interfaces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Binary nanoparticle (NP) superlattices exhibit distinct collective plasmonic, magnetic, optical, and electronic properties. Here, we computationally demonstrate how fluid-fluid interfaces could be used to self-assemble binary systems of NPs into 2D superlattices when the NP species exhibit different miscibility with the fluids forming the interface. We develop a basin-hopping Monte Carlo (BHMC) algorithm tailored for interface-trapped structures to rapidly determine the ground-state configuration of NPs, allowing us to explore the repertoire of binary NP architectures formed at the interface. By varying the NP size ratio, interparticle interaction strength, and difference in NP miscibility with the two fluids, we demonstrate the assembly of an array of exquisite 2D periodic architectures, including AB-, AB2-, and AB3-type monolayer superlattices as well as AB-, AB2-, A3B5-, and A4B6-type bilayer superlattices. Our results suggest that the interfacial assembly approach could be a versatile platform for fabricating 2D colloidal superlattices with tunable structure and properties. |
| 32. |  | Xueyan Chen, Qianqian Ding, Chao Bi, Jian Ruan, Shikuan Yang Lossless enrichment of trace analytes in levitating droplets for multiphase and multiplex detection In: Nature Communications, vol. 13, no. 7807, 2022, (Efficient enrichment of molecules from liquids, solid objects, or the gas phase is critical for their detection at trace concentrations. Here, the authors report on the lossless enrichment of analytes in droplets using acoustic levitation for multiphase and multiplex SERS detection.). @article{nokey,
title = {Lossless enrichment of trace analytes in levitating droplets for multiphase and multiplex detection},
author = {Xueyan Chen and Qianqian Ding and Chao Bi and Jian Ruan and Shikuan Yang},
url = {https://www.nature.com/articles/s41467-022-35495-9.pdf},
doi = {10.1038/s41467-022-35495-9},
year = {2022},
date = {2022-12-17},
urldate = {2022-12-17},
journal = {Nature Communications},
volume = {13},
number = {7807},
abstract = {Concentrating a trace amount of molecules from liquids, solid objects, or the gas phase and delivering them to a localized area are crucial for almost any trace analyte detection device. Analytes within a liquid droplet resting on micro/nanostructured surfaces with liquid-repellent coatings can be concentrated during solvent evaporation. However, these coatings suffer from complex manufacturing procedures, poor versatility, and limited analyte enrichment efficiency. Here, we report on the use of an acoustic levitation platform to losslessly concentrate the analyte molecules dissolved in any volatile liquid, attached to solid objects, or spread in air. Gold nanoparticles can be simultaneously concentrated with the analytes in different phases, realizing sensitive, surface-enhanced Raman scattering detection even at attomolar (10^−18 mol/L) concentration levels. The acoustic levitation platform-enabled, lossless analyte enrichment can significantly increase the analytical performance of many conventional microsensing techniques.},
note = {Efficient enrichment of molecules from liquids, solid objects, or the gas phase is critical for their detection at trace concentrations. Here, the authors report on the lossless enrichment of analytes in droplets using acoustic levitation for multiphase and multiplex SERS detection.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Concentrating a trace amount of molecules from liquids, solid objects, or the gas phase and delivering them to a localized area are crucial for almost any trace analyte detection device. Analytes within a liquid droplet resting on micro/nanostructured surfaces with liquid-repellent coatings can be concentrated during solvent evaporation. However, these coatings suffer from complex manufacturing procedures, poor versatility, and limited analyte enrichment efficiency. Here, we report on the use of an acoustic levitation platform to losslessly concentrate the analyte molecules dissolved in any volatile liquid, attached to solid objects, or spread in air. Gold nanoparticles can be simultaneously concentrated with the analytes in different phases, realizing sensitive, surface-enhanced Raman scattering detection even at attomolar (10^−18 mol/L) concentration levels. The acoustic levitation platform-enabled, lossless analyte enrichment can significantly increase the analytical performance of many conventional microsensing techniques. |
| 31. |  | Xinquan Zhou, Lixin Ning, Jianwei Qiao, Yifei Zhao, Puxian Xiong, Zhiguo Xia Interplay of defect levels and rare earth emission centers in multimode luminescent phosphors In: Nature Communications, vol. 13, no. 7589, 2022, (Information encryption technology calls for versatile multi-mode luminescent materials. Here, the authors develop phosphors with five integrated luminescence modes by exploiting the interplay of defect levels and rare-earth emission centers.). @article{nokey,
title = {Interplay of defect levels and rare earth emission centers in multimode luminescent phosphors},
author = {Xinquan Zhou and Lixin Ning and Jianwei Qiao and Yifei Zhao and Puxian Xiong and Zhiguo Xia },
url = {https://www.nature.com/articles/s41467-022-35366-3.pdf},
doi = {10.1038/s41467-022-35366-3},
year = {2022},
date = {2022-12-08},
urldate = {2022-12-08},
journal = {Nature Communications},
volume = {13},
number = {7589},
abstract = {Multimode luminescence generally involves tunable photon emissions in response to various excitation or stimuli channels, which demonstrates high coding capacity and confidentiality abilities for anti-counterfeiting and encryption technologies. Integrating multimode luminescence into a single stable material is a promising strategy but remains a challenge. Here, we realize distinct long persistent luminescence, short-lived down/upconversion emissions in NaGdTi2O6:Pr3+, Er3+ phosphor by emloying interplay of defect levels and rare earth emission centers. The materials show intense colorful luminescence statically and dynamically, which responds to a wide spectrum ranging from X-ray to sunlight, thermal disturbance, and mechanical force, further allowing the emission colors manipulable in space and time dimensions. Experimental and theoretical approaches reveal that the Pr3+ ↔ Pr4+ valence change, oxygen vacancies and anti-site TiGd defects in this disordered structure contributes to the multimode luminescence. We present a facile and nondestructive demo whose emission color and fade intensity can be controlled via external manipulation, indicating promise in high-capacity information encryption applications.},
note = {Information encryption technology calls for versatile multi-mode luminescent materials. Here, the authors develop phosphors with five integrated luminescence modes by exploiting the interplay of defect levels and rare-earth emission centers.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Multimode luminescence generally involves tunable photon emissions in response to various excitation or stimuli channels, which demonstrates high coding capacity and confidentiality abilities for anti-counterfeiting and encryption technologies. Integrating multimode luminescence into a single stable material is a promising strategy but remains a challenge. Here, we realize distinct long persistent luminescence, short-lived down/upconversion emissions in NaGdTi2O6:Pr3+, Er3+ phosphor by emloying interplay of defect levels and rare earth emission centers. The materials show intense colorful luminescence statically and dynamically, which responds to a wide spectrum ranging from X-ray to sunlight, thermal disturbance, and mechanical force, further allowing the emission colors manipulable in space and time dimensions. Experimental and theoretical approaches reveal that the Pr3+ ↔ Pr4+ valence change, oxygen vacancies and anti-site TiGd defects in this disordered structure contributes to the multimode luminescence. We present a facile and nondestructive demo whose emission color and fade intensity can be controlled via external manipulation, indicating promise in high-capacity information encryption applications. |
| 30. | 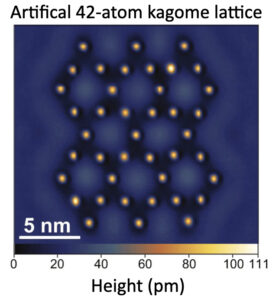 | I-Ju Chen, Markus Aapro, Abraham Kipnis, Alexander Ilin, Peter Liljeroth, Adam S. Foster Precise atom manipulation through deep reinforcement learning In: Nature Communications, vol. 13, no. 7499, 2022, (Engineering quantum states requires precise manipulations at the atomic level. Here, the authors use deep reinforcement learning to manipulate Ag adatoms on Ag surfaces, which combined with path planning algorithms enables autonomous atomic assembly.). @article{nokey,
title = {Precise atom manipulation through deep reinforcement learning},
author = {I-Ju Chen and Markus Aapro and Abraham Kipnis and Alexander Ilin and Peter Liljeroth and Adam S. Foster },
url = {https://www.nature.com/articles/s41467-022-35149-w.pdf},
doi = {10.1038/s41467-022-35149-w},
year = {2022},
date = {2022-12-05},
urldate = {2022-12-05},
journal = {Nature Communications},
volume = {13},
number = {7499},
abstract = {Atomic-scale manipulation in scanning tunneling microscopy has enabled the creation of quantum states of matter based on artificial structures and extreme miniaturization of computational circuitry based on individual atoms. The ability to autonomously arrange atomic structures with precision will enable the scaling up of nanoscale fabrication and expand the range of artificial structures hosting exotic quantum states. However, the a priori unknown manipulation parameters, the possibility of spontaneous tip apex changes, and the difficulty of modeling tip-atom interactions make it challenging to select manipulation parameters that can achieve atomic precision throughout extended operations. Here we use deep reinforcement learning (DRL) to control the real-world atom manipulation process. Several state-of-the-art reinforcement learning (RL) techniques are used jointly to boost data efficiency. The DRL agent learns to manipulate Ag adatoms on Ag(111) surfaces with optimal precision and is integrated with path planning algorithms to complete an autonomous atomic assembly system. The results demonstrate that state-of-the-art DRL can offer effective solutions to real-world challenges in nanofabrication and powerful approaches to increasingly complex scientific experiments at the atomic scale.},
note = {Engineering quantum states requires precise manipulations at the atomic level. Here, the authors use deep reinforcement learning to manipulate Ag adatoms on Ag surfaces, which combined with path planning algorithms enables autonomous atomic assembly.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Atomic-scale manipulation in scanning tunneling microscopy has enabled the creation of quantum states of matter based on artificial structures and extreme miniaturization of computational circuitry based on individual atoms. The ability to autonomously arrange atomic structures with precision will enable the scaling up of nanoscale fabrication and expand the range of artificial structures hosting exotic quantum states. However, the a priori unknown manipulation parameters, the possibility of spontaneous tip apex changes, and the difficulty of modeling tip-atom interactions make it challenging to select manipulation parameters that can achieve atomic precision throughout extended operations. Here we use deep reinforcement learning (DRL) to control the real-world atom manipulation process. Several state-of-the-art reinforcement learning (RL) techniques are used jointly to boost data efficiency. The DRL agent learns to manipulate Ag adatoms on Ag(111) surfaces with optimal precision and is integrated with path planning algorithms to complete an autonomous atomic assembly system. The results demonstrate that state-of-the-art DRL can offer effective solutions to real-world challenges in nanofabrication and powerful approaches to increasingly complex scientific experiments at the atomic scale. |
| 29. | 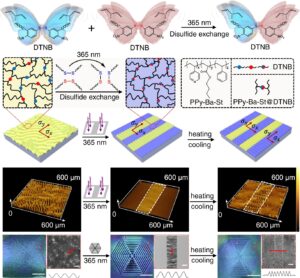 | Shuzhen Yan, Kaiming Hu, Shuai Chen, Tiantian Li, Wenming Zhang, Jie Yin, Xuesong Jiang Photo-induced stress relaxation in reconfigurable disulfide-crosslinked supramolecular films visualized by dynamic wrinkling In: Nature Communications, vol. 13, no. 7434, 2022, (The mechanics of reconfigurable supramolecular polymer networks are governed by their dynamic crosslinking chemistry and the resulting stress relaxations. Here, the authors use reversible wrinkling patterns to visualize localized stress relaxations, due to molecular network rearrangements.). @article{nokey,
title = {Photo-induced stress relaxation in reconfigurable disulfide-crosslinked supramolecular films visualized by dynamic wrinkling},
author = {Shuzhen Yan and Kaiming Hu and Shuai Chen and Tiantian Li and Wenming Zhang and Jie Yin and Xuesong Jiang },
url = {https://www.nature.com/articles/s41467-022-35271-9.pdf},
doi = {10.1038/s41467-022-35271-9},
year = {2022},
date = {2022-12-02},
urldate = {2022-12-02},
journal = {Nature Communications},
volume = {13},
number = {7434},
abstract = {Stress relaxation in reconfigurable supramolecular polymer networks is strongly related to intermolecular behavior. However, the relationship between molecular motion and macroscopic mechanics is usually vague, and the visualization of internal stress reflecting precise regulation of molecules remains challenging. Here, we present a strategy for visualizing photo-driven stress relaxation induced by infinitesimal perturbations in the intermolecular exchange reaction via reprogrammable wrinkle patterns. The supramolecular films exhibit visible changes in microscopic wrinkle topography through ultraviolet (UV)-induced dynamic disulfide exchange reaction. In accordance with the trans-scale theoretical models, which quantitatively evaluate the chemical-dependent mechanical stresses in the supramolecular network, the unexposed disordered wrinkles evolved into highly oriented patterns and underwent subsequent mutations after thermal treatment. The stress-sensitive wrinkle macro-patterns can be repetitively written/erased through network topology rearrangement using different stimuli. This strategy provides an approach for visualizing and understanding the molecular behavior from dynamic chemistry to mechanical changes, and directly programming wrinkle patterns with regulated structures.},
note = {The mechanics of reconfigurable supramolecular polymer networks are governed by their dynamic crosslinking chemistry and the resulting stress relaxations. Here, the authors use reversible wrinkling patterns to visualize localized stress relaxations, due to molecular network rearrangements.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Stress relaxation in reconfigurable supramolecular polymer networks is strongly related to intermolecular behavior. However, the relationship between molecular motion and macroscopic mechanics is usually vague, and the visualization of internal stress reflecting precise regulation of molecules remains challenging. Here, we present a strategy for visualizing photo-driven stress relaxation induced by infinitesimal perturbations in the intermolecular exchange reaction via reprogrammable wrinkle patterns. The supramolecular films exhibit visible changes in microscopic wrinkle topography through ultraviolet (UV)-induced dynamic disulfide exchange reaction. In accordance with the trans-scale theoretical models, which quantitatively evaluate the chemical-dependent mechanical stresses in the supramolecular network, the unexposed disordered wrinkles evolved into highly oriented patterns and underwent subsequent mutations after thermal treatment. The stress-sensitive wrinkle macro-patterns can be repetitively written/erased through network topology rearrangement using different stimuli. This strategy provides an approach for visualizing and understanding the molecular behavior from dynamic chemistry to mechanical changes, and directly programming wrinkle patterns with regulated structures. |
| 28. | 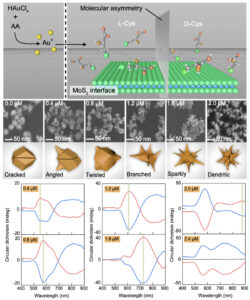 | Bang Lin Li, Jun Jiang Luo, Hao Lin Zou, Qing-Meng Zhang, Liu-Bin Zhao, Hang Qian, Hong Qun Luo, David Tai Leong, Nian Bing Li Chiral nanocrystals grown from MoS2 nanosheets enable photothermally modulated enantioselective release of antimicrobial drugs In: Nature Communications, vol. 13, no. 7289, 2022, (Chirality transfer from molecules to nanomaterials enables advanced optical functionalities. Here, the authors use exfoliated MoS2 nanosheets to seed the growth of chiral Au nanoparticles to form Au/MoS2 heterostructures for enantioselective drug release.). @article{nokey,
title = {Chiral nanocrystals grown from MoS2 nanosheets enable photothermally modulated enantioselective release of antimicrobial drugs},
author = {Bang Lin Li and Jun Jiang Luo and Hao Lin Zou and Qing-Meng Zhang and Liu-Bin Zhao and Hang Qian and Hong Qun Luo and David Tai Leong and Nian Bing Li },
url = {https://www.nature.com/articles/s41467-022-35016-8.pdf},
doi = {10.1038/s41467-022-35016-8},
year = {2022},
date = {2022-11-26},
urldate = {2022-11-26},
journal = {Nature Communications},
volume = {13},
number = {7289},
abstract = {The transfer of the concept of chirality from molecules to synthesized nanomaterials has attracted attention amongst multidisciplinary teams. Here we demonstrate heterogeneous nucleation and anisotropic accumulation of Au nanoparticles on multilayer MoS2 planes to form chiroptically functional nanomaterials. Thiol amino acids with chiral conformations modulate asymmetric growth of gold nanoarchitectures on seeds of highly faceted Au/MoS2 heterostructures. Consequently, dendritic plasmonic nanocrystals with partial chiral morphologies are synthesized. The chirality of dendritic nanocrystals inherited from cysteine molecules refers to the structural characteristics and includes specific recognition of enantiomeric molecules. With integration of the intrinsic photothermal properties and inherited enantioselective characteristics, dendritic Au/MoS2 heterostructures exhibit chirality-dependent release of antimicrobial drugs from hydrogel substrates when activated by exogenous infrared irradiation. A three-in-one strategy involving synthesis of chiral dendritic heterostructures, enantioselective recognition, and controlled drug release system is presented, which improves nanomaterial synthetic technology and enhances our understanding of crucial chirality information.},
note = {Chirality transfer from molecules to nanomaterials enables advanced optical functionalities. Here, the authors use exfoliated MoS2 nanosheets to seed the growth of chiral Au nanoparticles to form Au/MoS2 heterostructures for enantioselective drug release.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The transfer of the concept of chirality from molecules to synthesized nanomaterials has attracted attention amongst multidisciplinary teams. Here we demonstrate heterogeneous nucleation and anisotropic accumulation of Au nanoparticles on multilayer MoS2 planes to form chiroptically functional nanomaterials. Thiol amino acids with chiral conformations modulate asymmetric growth of gold nanoarchitectures on seeds of highly faceted Au/MoS2 heterostructures. Consequently, dendritic plasmonic nanocrystals with partial chiral morphologies are synthesized. The chirality of dendritic nanocrystals inherited from cysteine molecules refers to the structural characteristics and includes specific recognition of enantiomeric molecules. With integration of the intrinsic photothermal properties and inherited enantioselective characteristics, dendritic Au/MoS2 heterostructures exhibit chirality-dependent release of antimicrobial drugs from hydrogel substrates when activated by exogenous infrared irradiation. A three-in-one strategy involving synthesis of chiral dendritic heterostructures, enantioselective recognition, and controlled drug release system is presented, which improves nanomaterial synthetic technology and enhances our understanding of crucial chirality information. |
| 27. | 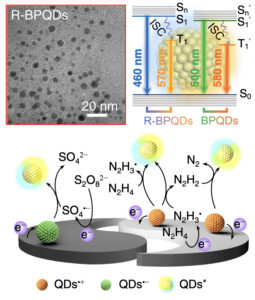 | Siqi Yu, Yu Du, Xianghong Niu, Guangming Li, Da Zhu, Qian Yu, Guizheng Zou, Huangxian Ju Arginine-modified black phosphorus quantum dots with dual excited states for enhanced electrochemiluminescence in bioanalysis In: Nature Communications, vol. 13, no. 7302, 2022, (Electrochemiluminescence is emitted via the radiative transition of a singlet or triplet excited state. Here, the authors propose an arginine modification of black phosphorus quantum dots that exhibits enhanced emission based on dual excited states.). @article{nokey,
title = {Arginine-modified black phosphorus quantum dots with dual excited states for enhanced electrochemiluminescence in bioanalysis},
author = {Siqi Yu and Yu Du and Xianghong Niu and Guangming Li and Da Zhu and Qian Yu and Guizheng Zou and Huangxian Ju},
url = {https://www.nature.com/articles/s41467-022-35015-9.pdf},
doi = {10.1038/s41467-022-35015-9},
year = {2022},
date = {2022-11-26},
urldate = {2022-11-26},
journal = {Nature Communications},
volume = {13},
number = {7302},
abstract = {The electrochemiluminescence (ECL) is generally emitted via radiative transition of singlet or triplet excited state (S1 or T1). Herein, an ECL mechanism with the transitions of both S1 and T1 of black phosphorus quantum dots (BPQDs) is found, and an arginine (Arg) modification strategy is proposed to passivate the surface oxidation defects of BPQDs, which could modulate the excited states for enhancing the ECL efficiency of BPQDs. The Arg modification leads to greater spatial overlap of highest and lowest occupied molecular orbitals, and spectral shift of radiative transitions, and improves the stability of anion radical of BPQDs. To verify the application of the proposed mechanism, it is used to construct a sensitive method for conveniently evaluating the inhibiting efficiency of cyclo-arginine-glycine-aspartic acid-d-tyrosine-lysine to cell surface integrin by using Arg containing peptide modified BPQDs as signal tag. The dual excited states mediated ECL emitters provide a paradigm for adjustable ECL generation and extend the application of ECL analysis.},
note = {Electrochemiluminescence is emitted via the radiative transition of a singlet or triplet excited state. Here, the authors propose an arginine modification of black phosphorus quantum dots that exhibits enhanced emission based on dual excited states.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The electrochemiluminescence (ECL) is generally emitted via radiative transition of singlet or triplet excited state (S1 or T1). Herein, an ECL mechanism with the transitions of both S1 and T1 of black phosphorus quantum dots (BPQDs) is found, and an arginine (Arg) modification strategy is proposed to passivate the surface oxidation defects of BPQDs, which could modulate the excited states for enhancing the ECL efficiency of BPQDs. The Arg modification leads to greater spatial overlap of highest and lowest occupied molecular orbitals, and spectral shift of radiative transitions, and improves the stability of anion radical of BPQDs. To verify the application of the proposed mechanism, it is used to construct a sensitive method for conveniently evaluating the inhibiting efficiency of cyclo-arginine-glycine-aspartic acid-d-tyrosine-lysine to cell surface integrin by using Arg containing peptide modified BPQDs as signal tag. The dual excited states mediated ECL emitters provide a paradigm for adjustable ECL generation and extend the application of ECL analysis. |
| 26. |  | Qingyu Meng, Laura Abella, Yang-Rong Yao, Dumitru-Claudiu Sergentu, Wei Yang, Xinye Liu, Jiaxin Zhuang, Luis Echegoyen, Jochen Autschbach, Ning Chen A charged diatomic triple-bonded U≡N species trapped in C82 fullerene cages In: Nature Communications, vol. 13, no. 7192, 2022, (Diatomic actinide molecules are ideal models for studying rare multiple-bond motifs. Here, the authors report host-guest structures of metastable charged U≡N diatoms confined in fullerene cages and stabilized by coordinative electron transfer.). @article{nokey,
title = {A charged diatomic triple-bonded U≡N species trapped in C82 fullerene cages},
author = {Qingyu Meng and Laura Abella and Yang-Rong Yao and Dumitru-Claudiu Sergentu and Wei Yang and Xinye Liu and Jiaxin Zhuang and Luis Echegoyen and Jochen Autschbach and Ning Chen },
url = {https://www.nature.com/articles/s41467-022-34651-5.pdf},
doi = {10.1038/s41467-022-34651-5},
year = {2022},
date = {2022-11-23},
urldate = {2022-11-23},
journal = {Nature Communications},
volume = {13},
number = {7192},
abstract = {Actinide diatomic molecules are ideal models to study elusive actinide multiple bonds, but most of these diatomic molecules have so far only been studied in solid inert gas matrices. Herein, we report a charged U≡N diatomic species captured in fullerene cages and stabilized by the U-fullerene coordination interaction. Two diatomic clusterfullerenes, viz. UN@Cs(6)-C82 and UN@C2(5)-C82, were successfully synthesized and characterized. Crystallographic analysis reveals U-N bond lengths of 1.760(7) and 1.760(20) Å in UN@Cs(6)-C82 and UN@C2(5)-C82. Moreover, U≡N was found to be immobilized and coordinated to the fullerene cages at 100 K but it rotates inside the cage at 273 K. Quantum-chemical calculations show a (UN)2+@(C82)2− electronic structure with formal +5 oxidation state (f1) of U and unambiguously demonstrate the presence of a U≡N bond in the clusterfullerenes. This study constitutes an approach to stabilize fundamentally important actinide multiply bonded species.},
note = {Diatomic actinide molecules are ideal models for studying rare multiple-bond motifs. Here, the authors report host-guest structures of metastable charged U≡N diatoms confined in fullerene cages and stabilized by coordinative electron transfer.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Actinide diatomic molecules are ideal models to study elusive actinide multiple bonds, but most of these diatomic molecules have so far only been studied in solid inert gas matrices. Herein, we report a charged U≡N diatomic species captured in fullerene cages and stabilized by the U-fullerene coordination interaction. Two diatomic clusterfullerenes, viz. UN@Cs(6)-C82 and UN@C2(5)-C82, were successfully synthesized and characterized. Crystallographic analysis reveals U-N bond lengths of 1.760(7) and 1.760(20) Å in UN@Cs(6)-C82 and UN@C2(5)-C82. Moreover, U≡N was found to be immobilized and coordinated to the fullerene cages at 100 K but it rotates inside the cage at 273 K. Quantum-chemical calculations show a (UN)2+@(C82)2− electronic structure with formal +5 oxidation state (f1) of U and unambiguously demonstrate the presence of a U≡N bond in the clusterfullerenes. This study constitutes an approach to stabilize fundamentally important actinide multiply bonded species. |
| 25. |  | Yuanzhi Xia, Semih Sevim, João Pedro Vale, Johannes Seibel, David Rodríguez-San-Miguel, Donghoon Kim, Salvador Pané, Tiago Sotto Mayor, Steven De Feyter, Josep Puigmartí-Luis Covalent transfer of chemical gradients onto a graphenic surface with 2D and 3D control In: Nature Communications, vol. 13, no. 7006, 2022, (Covalent modification is an essential chemical method for altering the physicochemical properties of material interfaces. Here, the authors show that the no-slip conditions in microfluidic devices grant spatiotemporal control over molecular grafting.). @article{nokey,
title = {Covalent transfer of chemical gradients onto a graphenic surface with 2D and 3D control},
author = {Yuanzhi Xia and Semih Sevim and João Pedro Vale and Johannes Seibel and David Rodríguez-San-Miguel and Donghoon Kim and Salvador Pané and Tiago Sotto Mayor and Steven De Feyter and Josep Puigmartí-Luis },
url = {https://www.nature.com/articles/s41467-022-34684-w.pdf},
doi = {10.1038/s41467-022-34684-w},
year = {2022},
date = {2022-11-16},
urldate = {2022-11-16},
journal = {Nature Communications},
volume = {13},
number = {7006},
abstract = {Control over the functionalization of graphenic materials is key to enable their full application in electronic and optical technologies. Covalent functionalization strategies have been proposed as an approach to tailor the interfaces’ structure and properties. However, to date, none of the proposed methods allow for a covalent functionalization with control over the grafting density, layer thickness and/or morphology, which are key aspects for fine-tuning the processability and performance of graphenic materials. Here, we show that the no-slip boundary condition at the walls of a continuous flow microfluidic device offers a way to generate controlled chemical gradients onto a graphenic material with 2D and 3D control, a possibility that will allow the sophisticated functionalization of these technologically-relevant materials.},
note = {Covalent modification is an essential chemical method for altering the physicochemical properties of material interfaces. Here, the authors show that the no-slip conditions in microfluidic devices grant spatiotemporal control over molecular grafting.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Control over the functionalization of graphenic materials is key to enable their full application in electronic and optical technologies. Covalent functionalization strategies have been proposed as an approach to tailor the interfaces’ structure and properties. However, to date, none of the proposed methods allow for a covalent functionalization with control over the grafting density, layer thickness and/or morphology, which are key aspects for fine-tuning the processability and performance of graphenic materials. Here, we show that the no-slip boundary condition at the walls of a continuous flow microfluidic device offers a way to generate controlled chemical gradients onto a graphenic material with 2D and 3D control, a possibility that will allow the sophisticated functionalization of these technologically-relevant materials. |
| 24. |  | Qianqian Fu, Wenyuan Yu, Guangyang Bao, Jianping Ge Electrically responsive photonic crystals with bistable states for low-power electrophoretic color displays In: Nature Communications, vol. 13, no. 7007, 2022, (Conventional electrophoretic color displays require a permanent electric field, increasing power consumption. Here, the authors report an electrically responsive photonic crystal with switchable bistable states by particle rearrangement for low power consumption displays.). @article{nokey,
title = {Electrically responsive photonic crystals with bistable states for low-power electrophoretic color displays},
author = {Qianqian Fu and Wenyuan Yu and Guangyang Bao and Jianping Ge },
url = {https://www.nature.com/articles/s41467-022-34745-0.pdf},
doi = {10.1038/s41467-022-34745-0},
year = {2022},
date = {2022-11-16},
urldate = {2022-11-16},
journal = {Nature Communications},
volume = {13},
number = {7007},
abstract = {Electrically responsive photonic crystals are promising materials for electrophoretic color displays with better brightness and color saturation. However, electric field must always be applied to maintain the specific colors, which brings concerns about the power consumption and signal stability and reversibility. Here, we show an electrically responsive photonic crystal with two stable states at 0 V, which are the colored state or the colorless state with ordered or disordered particle arrangement. The color state can be reversibly switched by applying a short-time electrical field, just like in the case of commercial electrophoretic ink. With optimized recipe and electric field, the photonic crystals encapsulated in the prototype display panel are proved to have potentials in high resolution, multi-color, and greyscale display, which lays down a firm basis for reflective displays with low power consumption and good visibility.},
note = {Conventional electrophoretic color displays require a permanent electric field, increasing power consumption. Here, the authors report an electrically responsive photonic crystal with switchable bistable states by particle rearrangement for low power consumption displays.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Electrically responsive photonic crystals are promising materials for electrophoretic color displays with better brightness and color saturation. However, electric field must always be applied to maintain the specific colors, which brings concerns about the power consumption and signal stability and reversibility. Here, we show an electrically responsive photonic crystal with two stable states at 0 V, which are the colored state or the colorless state with ordered or disordered particle arrangement. The color state can be reversibly switched by applying a short-time electrical field, just like in the case of commercial electrophoretic ink. With optimized recipe and electric field, the photonic crystals encapsulated in the prototype display panel are proved to have potentials in high resolution, multi-color, and greyscale display, which lays down a firm basis for reflective displays with low power consumption and good visibility. |
| 23. |  | Ahyoung Kim, Thi Vo, Hyosung An, Progna Banerjee, Lehan Yao, Shan Zhou, Chansong Kim, Delia J. Milliron, Sharon C. Glotzer, Qian Chen Symmetry-breaking in patch formation on triangular gold nanoparticles by asymmetric polymer grafting In: Nature Communications, vol. 13, no. 6774, 2022, (Patchy nanoparticles are desirable building blocks for the guided assembly of functional superstructures. Here, the authors demonstrate quantitative control over asymmetric polymer grafting on triangular Au nanoprisms based on polymer scaling theory.). @article{nokey,
title = {Symmetry-breaking in patch formation on triangular gold nanoparticles by asymmetric polymer grafting},
author = {Ahyoung Kim and Thi Vo and Hyosung An and Progna Banerjee and Lehan Yao and Shan Zhou and Chansong Kim and Delia J. Milliron and Sharon C. Glotzer and Qian Chen },
url = {https://www.nature.com/articles/s41467-022-34246-0.pdf},
doi = {10.1038/s41467-022-34246-0},
year = {2022},
date = {2022-11-09},
urldate = {2022-11-09},
journal = {Nature Communications},
volume = {13},
number = {6774},
abstract = {Synthesizing patchy particles with predictive control over patch size, shape, placement and number has been highly sought-after for nanoparticle assembly research, but is fraught with challenges. Here we show that polymers can be designed to selectively adsorb onto nanoparticle surfaces already partially coated by other chains to drive the formation of patchy nanoparticles with broken symmetry. In our model system of triangular gold nanoparticles and polystyrene-b-polyacrylic acid patch, single- and double-patch nanoparticles are produced at high yield. These asymmetric single-patch nanoparticles are shown to assemble into self-limited patch‒patch connected bowties exhibiting intriguing plasmonic properties. To unveil the mechanism of symmetry-breaking patch formation, we develop a theory that accurately predicts our experimental observations at all scales—from patch patterning on nanoparticles, to the size/shape of the patches, to the particle assemblies driven by patch‒patch interactions. Both the experimental strategy and theoretical prediction extend to nanoparticles of other shapes such as octahedra and bipyramids. Our work provides an approach to leverage polymer interactions with nanoscale curved surfaces for asymmetric grafting in nanomaterials engineering.},
note = {Patchy nanoparticles are desirable building blocks for the guided assembly of functional superstructures. Here, the authors demonstrate quantitative control over asymmetric polymer grafting on triangular Au nanoprisms based on polymer scaling theory.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Synthesizing patchy particles with predictive control over patch size, shape, placement and number has been highly sought-after for nanoparticle assembly research, but is fraught with challenges. Here we show that polymers can be designed to selectively adsorb onto nanoparticle surfaces already partially coated by other chains to drive the formation of patchy nanoparticles with broken symmetry. In our model system of triangular gold nanoparticles and polystyrene-b-polyacrylic acid patch, single- and double-patch nanoparticles are produced at high yield. These asymmetric single-patch nanoparticles are shown to assemble into self-limited patch‒patch connected bowties exhibiting intriguing plasmonic properties. To unveil the mechanism of symmetry-breaking patch formation, we develop a theory that accurately predicts our experimental observations at all scales—from patch patterning on nanoparticles, to the size/shape of the patches, to the particle assemblies driven by patch‒patch interactions. Both the experimental strategy and theoretical prediction extend to nanoparticles of other shapes such as octahedra and bipyramids. Our work provides an approach to leverage polymer interactions with nanoscale curved surfaces for asymmetric grafting in nanomaterials engineering. |
| 22. |  | Pingping Zhang, Gaoling Yang, Fei Li, Jianbing Shi, Haizheng Zhong Direct in situ photolithography of perovskite quantum dots based on photocatalysis of lead bromide complexes In: Nature Communications, vol. 13, no. 6713, 2022, (Perovskite nanomaterials may suffer degradation during conventional photolithography. Here, the authors report a non-destructive method for patterning perovskite quantum dots based on direct photopolymerization catalyzed by lead bromide complexes.). @article{nokey,
title = {Direct in situ photolithography of perovskite quantum dots based on photocatalysis of lead bromide complexes},
author = {Pingping Zhang and Gaoling Yang and Fei Li and Jianbing Shi and Haizheng Zhong },
url = {https://www.nature.com/articles/s41467-022-34453-9.pdf},
doi = {10.1038/s41467-022-34453-9},
year = {2022},
date = {2022-11-07},
urldate = {2022-11-07},
journal = {Nature Communications},
volume = {13},
number = {6713},
abstract = {Photolithography has shown great potential in patterning solution-processed nanomaterials for integration into advanced optoelectronic devices. However, photolithography of perovskite quantum dots (PQDs) has so far been hindered by the incompatibility of perovskite with traditional optical lithography processes where lots of solvents and high-energy ultraviolet (UV) light exposure are required. Herein, we report a direct in situ photolithography technique to pattern PQDs based on the photopolymerization catalyzed by lead bromide complexes. By combining direct photolithography with in situ fabrication of PQDs, this method allows to directly photolithograph perovskite precursors, avoiding the complicated lift-off processes and the destruction of PQDs by solvents or high-energy UV light, as PQDs are produced after lithography exposure. We further demonstrate that the thiol-ene free-radical photopolymerization is catalyzed by lead bromide complexes in the perovskite precursor solution, while no external initiators or catalysts are needed. Using direct in situ photolithography, PQD patterns with high resolution up to 2450 pixels per inch (PPI), excellent fluorescence uniformity, and good stability, are successfully demonstrated. This work opens an avenue for non-destructive direct photolithography of high-efficiency light-emitting PQDs, and potentially expands their application in various integrated optoelectronic devices.},
note = {Perovskite nanomaterials may suffer degradation during conventional photolithography. Here, the authors report a non-destructive method for patterning perovskite quantum dots based on direct photopolymerization catalyzed by lead bromide complexes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Photolithography has shown great potential in patterning solution-processed nanomaterials for integration into advanced optoelectronic devices. However, photolithography of perovskite quantum dots (PQDs) has so far been hindered by the incompatibility of perovskite with traditional optical lithography processes where lots of solvents and high-energy ultraviolet (UV) light exposure are required. Herein, we report a direct in situ photolithography technique to pattern PQDs based on the photopolymerization catalyzed by lead bromide complexes. By combining direct photolithography with in situ fabrication of PQDs, this method allows to directly photolithograph perovskite precursors, avoiding the complicated lift-off processes and the destruction of PQDs by solvents or high-energy UV light, as PQDs are produced after lithography exposure. We further demonstrate that the thiol-ene free-radical photopolymerization is catalyzed by lead bromide complexes in the perovskite precursor solution, while no external initiators or catalysts are needed. Using direct in situ photolithography, PQD patterns with high resolution up to 2450 pixels per inch (PPI), excellent fluorescence uniformity, and good stability, are successfully demonstrated. This work opens an avenue for non-destructive direct photolithography of high-efficiency light-emitting PQDs, and potentially expands their application in various integrated optoelectronic devices. |
| 21. |  | Tolulope Michael Ajayi, Vijay Singh, Kyaw Zin Latt, Sanjoy Sarkar, Xinyue Cheng, Sineth Premarathna, Naveen K. Dandu, Shaoze Wang, Fahimeh Movahedifar, Sarah Wieghold, Nozomi Shirato, Volker Rose, Larry A. Curtiss, Anh T. Ngo, Eric Masson, Saw Wai Hla Atomically precise control of rotational dynamics in charged rare-earth complexes on a metal surface In: Nature Communications, vol. 13, no. 6305, 2022, (Rare-earth elements are vital to advanced technological applications ranging from spintronic devices to quantum information science. Here, the authors formed charged rare-earth complexes on a material surface and demonstrated atomically precise control on their rotational dynamics.). @article{nokey,
title = {Atomically precise control of rotational dynamics in charged rare-earth complexes on a metal surface},
author = {Tolulope Michael Ajayi and Vijay Singh and Kyaw Zin Latt and Sanjoy Sarkar and Xinyue Cheng and Sineth Premarathna and Naveen K. Dandu and Shaoze Wang and Fahimeh Movahedifar and Sarah Wieghold and Nozomi Shirato and Volker Rose and Larry A. Curtiss and Anh T. Ngo and Eric Masson and Saw Wai Hla },
url = {https://www.nature.com/articles/s41467-022-33897-3.pdf},
doi = {10.1038/s41467-022-33897-3},
year = {2022},
date = {2022-10-22},
urldate = {2022-10-22},
journal = {Nature Communications},
volume = {13},
number = {6305},
abstract = {Complexes containing rare-earth ions attract great attention for their technological applications ranging from spintronic devices to quantum information science. While charged rare-earth coordination complexes are ubiquitous in solution, they are challenging to form on materials surfaces that would allow investigations for potential solid-state applications. Here we report formation and atomically precise manipulation of rare-earth complexes on a gold surface. Although they are composed of multiple units held together by electrostatic interactions, the entire complex rotates as a single unit when electrical energy is supplied from a scanning tunneling microscope tip. Despite the hexagonal symmetry of the gold surface, a counterion at the side of the complex guides precise three-fold rotations and 100% control of their rotational directions is achieved using a negative electric field from the scanning probe tip. This work demonstrates that counterions can be used to control dynamics of rare-earth complexes on materials surfaces for quantum and nanomechanical applications.},
note = {Rare-earth elements are vital to advanced technological applications ranging from spintronic devices to quantum information science. Here, the authors formed charged rare-earth complexes on a material surface and demonstrated atomically precise control on their rotational dynamics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Complexes containing rare-earth ions attract great attention for their technological applications ranging from spintronic devices to quantum information science. While charged rare-earth coordination complexes are ubiquitous in solution, they are challenging to form on materials surfaces that would allow investigations for potential solid-state applications. Here we report formation and atomically precise manipulation of rare-earth complexes on a gold surface. Although they are composed of multiple units held together by electrostatic interactions, the entire complex rotates as a single unit when electrical energy is supplied from a scanning tunneling microscope tip. Despite the hexagonal symmetry of the gold surface, a counterion at the side of the complex guides precise three-fold rotations and 100% control of their rotational directions is achieved using a negative electric field from the scanning probe tip. This work demonstrates that counterions can be used to control dynamics of rare-earth complexes on materials surfaces for quantum and nanomechanical applications. |
| 20. |  | Yuzhu Ma, Hongjin Zhang, Runfeng Lin, Yan Ai, Kun Lan, Linlin Duan, Wenyao Chen, Xuezhi Duan, Bing Ma, Changyao Wang, Xiaomin Li, Dongyuan Zhao Remodeling nanodroplets into hierarchical mesoporous silica nanoreactors with multiple chambers In: Nature Communications, vol. 13, no. 6136, 2022, (Multi-chambered structures have attracted great attention due to their ability to create multifunctional partitions in different chambers. Here, the authors prepared mesoporous silica nanoreactors with hierarchical chambers for catalytic cascades.). @article{nokey,
title = {Remodeling nanodroplets into hierarchical mesoporous silica nanoreactors with multiple chambers},
author = {Yuzhu Ma and Hongjin Zhang and Runfeng Lin and Yan Ai and Kun Lan and Linlin Duan and Wenyao Chen and Xuezhi Duan and Bing Ma and Changyao Wang and Xiaomin Li and Dongyuan Zhao },
url = {https://www.nature.com/articles/s41467-022-33856-y.pdf},
doi = {10.1038/s41467-022-33856-y},
year = {2022},
date = {2022-10-17},
urldate = {2022-10-17},
journal = {Nature Communications},
volume = {13},
number = {6136},
abstract = {Multi-chambered architectures have attracted much attention due to the ability to establish multifunctional partitions in different chambers, but manipulating the chamber numbers and coupling multi-functionality within the multi-chambered mesoporous nanoparticle remains a challenge. Herein, we propose a nanodroplet remodeling strategy for the synthesis of hierarchical multi-chambered mesoporous silica nanoparticles with tunable architectures. Typically, the dual-chambered nanoparticles with a high surface area of ~469 m2 g−1 present two interconnected cavities like a calabash. Furthermore, based on this nanodroplet remodeling strategy, multiple species (magnetic, catalytic, optic, etc.) can be separately anchored in different chamber without obvious mutual-crosstalk. We design a dual-chambered mesoporous nanoreactors with spatial isolation of Au and Pd active-sites for the cascade synthesis of 2-phenylindole from 1-nitro-2-(phenylethynyl)benzene. Due to the efficient mass transfer of reactants and intermediates in the dual-chambered structure, the selectivity of the target product reaches to ~76.5%, far exceeding that of single-chambered nanoreactors (~41.3%).},
note = {Multi-chambered structures have attracted great attention due to their ability to create multifunctional partitions in different chambers. Here, the authors prepared mesoporous silica nanoreactors with hierarchical chambers for catalytic cascades.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Multi-chambered architectures have attracted much attention due to the ability to establish multifunctional partitions in different chambers, but manipulating the chamber numbers and coupling multi-functionality within the multi-chambered mesoporous nanoparticle remains a challenge. Herein, we propose a nanodroplet remodeling strategy for the synthesis of hierarchical multi-chambered mesoporous silica nanoparticles with tunable architectures. Typically, the dual-chambered nanoparticles with a high surface area of ~469 m2 g−1 present two interconnected cavities like a calabash. Furthermore, based on this nanodroplet remodeling strategy, multiple species (magnetic, catalytic, optic, etc.) can be separately anchored in different chamber without obvious mutual-crosstalk. We design a dual-chambered mesoporous nanoreactors with spatial isolation of Au and Pd active-sites for the cascade synthesis of 2-phenylindole from 1-nitro-2-(phenylethynyl)benzene. Due to the efficient mass transfer of reactants and intermediates in the dual-chambered structure, the selectivity of the target product reaches to ~76.5%, far exceeding that of single-chambered nanoreactors (~41.3%). |
| 19. |  | Yixuan Gao, Li Huang, Yun Cao, Marcus Richter, Jing Qi, Qi Zheng, Huan Yang, Ji Ma, Xiao Chang, Xiaoshuai Fu, Carlos-Andres Palma, Hongliang Lu, Yu-Yang Zhang, Zhihai Cheng, Xiao Lin, Min Ouyang, Xinliang Feng, Shixuan Du, Hong-Jun Gao Selective activation of four quasi-equivalent C–H bonds yields N-doped graphene nanoribbons with partial corannulene motifs In: Nature Communications, vol. 13, no. 6146, 2022, (Selective activation of C–H bonds is a key challenge in organic reactions. Here, the authors achieve the selective activation of four quasi-equivalent C–H bonds, leading to the formation of N-doped graphene nanoribbons with partial corannulene motifs.). @article{nokey,
title = {Selective activation of four quasi-equivalent C–H bonds yields N-doped graphene nanoribbons with partial corannulene motifs},
author = {Yixuan Gao and Li Huang and Yun Cao and Marcus Richter and Jing Qi and Qi Zheng and Huan Yang and Ji Ma and Xiao Chang and Xiaoshuai Fu and Carlos-Andres Palma and Hongliang Lu and Yu-Yang Zhang and Zhihai Cheng and Xiao Lin and Min Ouyang and Xinliang Feng and Shixuan Du and Hong-Jun Gao },
url = {https://www.nature.com/articles/s41467-022-33898-2.pdf},
doi = {10.1038/s41467-022-33898-2},
year = {2022},
date = {2022-10-17},
urldate = {2022-10-17},
journal = {Nature Communications},
volume = {13},
number = {6146},
abstract = {Selective C–H bond activation is one of the most challenging topics for organic reactions. The difficulties arise not only from the high C–H bond dissociation enthalpies but also the existence of multiple equivalent/quasi-equivalent reaction sites in organic molecules. Here, we successfully achieve the selective activation of four quasi-equivalent C–H bonds in a specially designed nitrogen-containing polycyclic hydrocarbon (N-PH). Density functional theory calculations reveal that the adsorption of N-PH on Ag(100) differentiates the activity of the four ortho C(sp3) atoms in the N-heterocycles into two groups, suggesting a selective dehydrogenation, which is demonstrated by sequential-annealing experiments of N-PH/Ag(100). Further annealing leads to the formation of N-doped graphene nanoribbons with partial corannulene motifs, realized by the C–H bond activation process. Our work provides a route of designing precursor molecules with ortho C(sp3) atom in an N-heterocycle to realize surface-induced selective dehydrogenation in quasi-equivalent sites.},
note = {Selective activation of C–H bonds is a key challenge in organic reactions. Here, the authors achieve the selective activation of four quasi-equivalent C–H bonds, leading to the formation of N-doped graphene nanoribbons with partial corannulene motifs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Selective C–H bond activation is one of the most challenging topics for organic reactions. The difficulties arise not only from the high C–H bond dissociation enthalpies but also the existence of multiple equivalent/quasi-equivalent reaction sites in organic molecules. Here, we successfully achieve the selective activation of four quasi-equivalent C–H bonds in a specially designed nitrogen-containing polycyclic hydrocarbon (N-PH). Density functional theory calculations reveal that the adsorption of N-PH on Ag(100) differentiates the activity of the four ortho C(sp3) atoms in the N-heterocycles into two groups, suggesting a selective dehydrogenation, which is demonstrated by sequential-annealing experiments of N-PH/Ag(100). Further annealing leads to the formation of N-doped graphene nanoribbons with partial corannulene motifs, realized by the C–H bond activation process. Our work provides a route of designing precursor molecules with ortho C(sp3) atom in an N-heterocycle to realize surface-induced selective dehydrogenation in quasi-equivalent sites. |
| 18. |  | Chong Li, Qi Liu, Shengyang Tao Coemissive luminescent nanoparticles combining aggregation-induced emission and quenching dyes prepared in continuous flow In: Nature Communications, vol. 13, no. 6034, 2022, (Developing efficient light harvesting systems at low cost is a challenge. Here, the authors synthesized coemissive dyes in a continuous flow microreactor featuring a controlled cascade FRET process combining aggregation-induced emission and quenching.). @article{nokey,
title = {Coemissive luminescent nanoparticles combining aggregation-induced emission and quenching dyes prepared in continuous flow},
author = {Chong Li and Qi Liu and Shengyang Tao },
url = {https://www.nature.com/articles/s41467-022-33857-x.pdf},
doi = {10.1038/s41467-022-33857-x},
year = {2022},
date = {2022-10-13},
urldate = {2022-10-13},
journal = {Nature Communications},
volume = {13},
number = {6034},
abstract = {Achieving an ideal light-harvesting system at a low cost remains a challenge. Herein, we report the synthesis of a hybrid dye system based on tetraphenylene (TPE) encapsulated organic dyes in a continuous flow microreactor. The composite dye nanoparticles (NPs) are synthesized based on supramolecular self-assembly to achieve the co-emission of aggregation-induced emission dyes and aggregation-caused quenching dyes (CEAA). Numerical simulations and molecular spectroscopy were used to investigate the synthesis mechanism of the CEAA dyes. Nanoparticles of CEAA dyes provide a platform for efficient cascade Förster resonance energy transfer (FRET). Composite dye nanoparticles of TPE and Nile red (NiR) are synthesized for an ideal light-harvesting system using coumarin 6 (C-6) as an energy intermediate. The light-harvesting system has a considerable red-shift distance (~126 nm), high energy-transfer efficiency (ΦET) of 99.37%, and an antenna effect of 26.23. Finally, the versatility of the preparation method and the diversity of CEAA dyes are demonstrated.},
note = {Developing efficient light harvesting systems at low cost is a challenge. Here, the authors synthesized coemissive dyes in a continuous flow microreactor featuring a controlled cascade FRET process combining aggregation-induced emission and quenching.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Achieving an ideal light-harvesting system at a low cost remains a challenge. Herein, we report the synthesis of a hybrid dye system based on tetraphenylene (TPE) encapsulated organic dyes in a continuous flow microreactor. The composite dye nanoparticles (NPs) are synthesized based on supramolecular self-assembly to achieve the co-emission of aggregation-induced emission dyes and aggregation-caused quenching dyes (CEAA). Numerical simulations and molecular spectroscopy were used to investigate the synthesis mechanism of the CEAA dyes. Nanoparticles of CEAA dyes provide a platform for efficient cascade Förster resonance energy transfer (FRET). Composite dye nanoparticles of TPE and Nile red (NiR) are synthesized for an ideal light-harvesting system using coumarin 6 (C-6) as an energy intermediate. The light-harvesting system has a considerable red-shift distance (~126 nm), high energy-transfer efficiency (ΦET) of 99.37%, and an antenna effect of 26.23. Finally, the versatility of the preparation method and the diversity of CEAA dyes are demonstrated. |
| 17. |  | Jiří Doležal, Sofia Canola, Prokop Hapala, Rodrigo Cezar de Campos Ferreira, Pablo Merino, Martin Švec Evidence of exciton-libron coupling in chirally adsorbed single molecules In: Nature Communications, vol. 13, no. 6008, 2022, (Vibronic coupling in molecules plays an essential role in photophysics. Here, the authors observe optical fingerprints of the coupling between librational states and charged excited states in a single phthalocyanine molecule chirally absorbed on a surface.). @article{nokey,
title = {Evidence of exciton-libron coupling in chirally adsorbed single molecules},
author = {Jiří Doležal and Sofia Canola and Prokop Hapala and Rodrigo Cezar de Campos Ferreira and Pablo Merino and Martin Švec },
url = {https://www.nature.com/articles/s41467-022-33653-7.pdf},
doi = {10.1038/s41467-022-33653-7},
year = {2022},
date = {2022-10-12},
urldate = {2022-10-12},
journal = {Nature Communications},
volume = {13},
number = {6008},
abstract = {Interplay between motion of nuclei and excitations has an important role in molecular photophysics of natural and artificial structures. Here we provide a detailed analysis of coupling between quantized librational modes (librons) and charged excited states (trions) on single phthalocyanine dyes adsorbed on a surface. By means of tip-induced electroluminescence performed with a scanning probe microscope, we identify libronic signatures in spectra of chirally adsorbed phthalocyanines and find that these signatures are absent from spectra of symmetrically adsorbed species. We create a model of the libronic coupling based on the Franck-Condon principle to simulate the spectral features. Experimentally measured librational spectra match very well the theoretically calculated librational eigenenergies and peak intensities (Franck-Condon factors). Moreover, the comparison reveals an unexpected depopulation channel for the zero libron of the excited state that can be effectively controlled by tuning the size of the nanocavity. Our results showcase the possibility of characterizing the dynamics of molecules by their low-energy molecular modes using µeV-resolved tip-enhanced spectroscopy.},
note = {Vibronic coupling in molecules plays an essential role in photophysics. Here, the authors observe optical fingerprints of the coupling between librational states and charged excited states in a single phthalocyanine molecule chirally absorbed on a surface.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Interplay between motion of nuclei and excitations has an important role in molecular photophysics of natural and artificial structures. Here we provide a detailed analysis of coupling between quantized librational modes (librons) and charged excited states (trions) on single phthalocyanine dyes adsorbed on a surface. By means of tip-induced electroluminescence performed with a scanning probe microscope, we identify libronic signatures in spectra of chirally adsorbed phthalocyanines and find that these signatures are absent from spectra of symmetrically adsorbed species. We create a model of the libronic coupling based on the Franck-Condon principle to simulate the spectral features. Experimentally measured librational spectra match very well the theoretically calculated librational eigenenergies and peak intensities (Franck-Condon factors). Moreover, the comparison reveals an unexpected depopulation channel for the zero libron of the excited state that can be effectively controlled by tuning the size of the nanocavity. Our results showcase the possibility of characterizing the dynamics of molecules by their low-energy molecular modes using µeV-resolved tip-enhanced spectroscopy. |
| 16. |  | Arrigo Calzolari, Corey Oses, Cormac Toher, Marco Esters, Xiomara Campilongo, Sergei P. Stepanoff, Douglas E. Wolfe, Stefano Curtarolo Plasmonic high-entropy carbides In: Nature Communications, vol. 13, no. 5993, 2022, (Tunable plasmonic materials capable of surviving harsh environments are critical for advanced applications. Here, the authors report that some high-entropy transition-metal carbides can satisfy the requirements.). @article{nokey,
title = {Plasmonic high-entropy carbides},
author = {Arrigo Calzolari and Corey Oses and Cormac Toher and Marco Esters and Xiomara Campilongo and Sergei P. Stepanoff and Douglas E. Wolfe and Stefano Curtarolo },
url = {https://www.nature.com/articles/s41467-022-33497-1.pdf},
doi = {10.1038/s41467-022-33497-1},
year = {2022},
date = {2022-10-11},
urldate = {2022-10-11},
journal = {Nature Communications},
volume = {13},
number = {5993},
abstract = {Discovering multifunctional materials with tunable plasmonic properties, capable of surviving harsh environments is critical for advanced optical and telecommunication applications. We chose high-entropy transition-metal carbides because of their exceptional thermal, chemical stability, and mechanical properties. By integrating computational thermodynamic disorder modeling and time-dependent density functional theory characterization, we discovered a crossover energy in the infrared and visible range, corresponding to a metal-to-dielectric transition, exploitable for plasmonics. It was also found that the optical response of high-entropy carbides can be largely tuned from the near-IR to visible when changing the transition metal components and their concentration. By monitoring the electronic structures, we suggest rules for optimizing optical properties and designing tailored high-entropy ceramics. Experiments performed on the archetype carbide HfTa4C5 yielded plasmonic properties from room temperature to 1500K. Here we propose plasmonic transition-metal high-entropy carbides as a class of multifunctional materials. Their combination of plasmonic activity, high-hardness, and extraordinary thermal stability will result in yet unexplored applications.},
note = {Tunable plasmonic materials capable of surviving harsh environments are critical for advanced applications. Here, the authors report that some high-entropy transition-metal carbides can satisfy the requirements.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Discovering multifunctional materials with tunable plasmonic properties, capable of surviving harsh environments is critical for advanced optical and telecommunication applications. We chose high-entropy transition-metal carbides because of their exceptional thermal, chemical stability, and mechanical properties. By integrating computational thermodynamic disorder modeling and time-dependent density functional theory characterization, we discovered a crossover energy in the infrared and visible range, corresponding to a metal-to-dielectric transition, exploitable for plasmonics. It was also found that the optical response of high-entropy carbides can be largely tuned from the near-IR to visible when changing the transition metal components and their concentration. By monitoring the electronic structures, we suggest rules for optimizing optical properties and designing tailored high-entropy ceramics. Experiments performed on the archetype carbide HfTa4C5 yielded plasmonic properties from room temperature to 1500K. Here we propose plasmonic transition-metal high-entropy carbides as a class of multifunctional materials. Their combination of plasmonic activity, high-hardness, and extraordinary thermal stability will result in yet unexplored applications. |
| 15. |  | Hyesung Jo, Dae Han Wi, Taegu Lee, Yongmin Kwon, Chaehwa Jeong, Juhyeok Lee, Hionsuck Baik, Alexander J. Pattison, Wolfgang Theis, Colin Ophus, Peter Ercius, Yea-Lee Lee, Seunghwa Ryu, Sang Woo Han, Yongsoo Yang Direct strain correlations at the single-atom level in three-dimensional core-shell interface structures In: Nature Communications, vol. 13, no. 5957, 2022, (Understanding 3D interfacial strain at the atomic level has been a long-sought challenge in the field of core-shell nanomaterials. Here, the authors address this challenge by revealing the full 3D atomic structures of Pd@Pt core-shell nanoparticles.). @article{nokey,
title = {Direct strain correlations at the single-atom level in three-dimensional core-shell interface structures},
author = {Hyesung Jo and Dae Han Wi and Taegu Lee and Yongmin Kwon and Chaehwa Jeong and Juhyeok Lee and Hionsuck Baik and Alexander J. Pattison and Wolfgang Theis and Colin Ophus and Peter Ercius and Yea-Lee Lee and Seunghwa Ryu and Sang Woo Han and Yongsoo Yang },
url = {https://www.nature.com/articles/s41467-022-33236-6.pdf},
doi = {10.1038/s41467-022-33236-6},
year = {2022},
date = {2022-10-10},
urldate = {2022-10-10},
journal = {Nature Communications},
volume = {13},
number = {5957},
abstract = {Nanomaterials with core-shell architectures are prominent examples of strain-engineered materials. The lattice mismatch between the core and shell materials can cause strong interface strain, which affects the surface structures. Therefore, surface functional properties such as catalytic activities can be designed by fine-tuning the misfit strain at the interface. To precisely control the core-shell effect, it is essential to understand how the surface and interface strains are related at the atomic scale. Here, we elucidate the surface-interface strain relations by determining the full 3D atomic structure of Pd@Pt core-shell nanoparticles at the single-atom level via atomic electron tomography. Full 3D displacement fields and strain profiles of core-shell nanoparticles were obtained, which revealed a direct correlation between the surface and interface strain. The strain distributions show a strong shape-dependent anisotropy, whose nature was further corroborated by molecular statics simulations. From the observed surface strains, the surface oxygen reduction reaction activities were predicted. These findings give a deep understanding of structure-property relationships in strain-engineerable core-shell systems, which can lead to direct control over the resulting catalytic properties.},
note = {Understanding 3D interfacial strain at the atomic level has been a long-sought challenge in the field of core-shell nanomaterials. Here, the authors address this challenge by revealing the full 3D atomic structures of Pd@Pt core-shell nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanomaterials with core-shell architectures are prominent examples of strain-engineered materials. The lattice mismatch between the core and shell materials can cause strong interface strain, which affects the surface structures. Therefore, surface functional properties such as catalytic activities can be designed by fine-tuning the misfit strain at the interface. To precisely control the core-shell effect, it is essential to understand how the surface and interface strains are related at the atomic scale. Here, we elucidate the surface-interface strain relations by determining the full 3D atomic structure of Pd@Pt core-shell nanoparticles at the single-atom level via atomic electron tomography. Full 3D displacement fields and strain profiles of core-shell nanoparticles were obtained, which revealed a direct correlation between the surface and interface strain. The strain distributions show a strong shape-dependent anisotropy, whose nature was further corroborated by molecular statics simulations. From the observed surface strains, the surface oxygen reduction reaction activities were predicted. These findings give a deep understanding of structure-property relationships in strain-engineerable core-shell systems, which can lead to direct control over the resulting catalytic properties. |
| 14. | 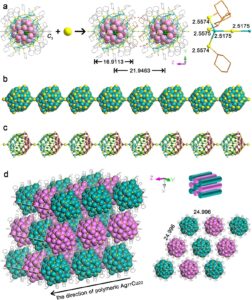 | Nan Xia, Jianpei Xing, Di Peng, Shiyu Ji, Jun Zha, Nan Yan, Yan Su, Xue Jiang, Zhi Zeng, Jijun Zhao, Zhikun Wu
Assembly-induced spin transfer and distance-dependent spin coupling in atomically precise AgCu nanoclusters In: Nature Communications, vol. 13, no. 5934, 2022, (The assembly of atomically precise clusters into ordered superstructures enables new functional material designs. Here, the authors propose a strategy for linear arrangements of AgCu clusters and explore the consequent transfer and coupling of magnetic spins.). @article{nokey,
title = {Assembly-induced spin transfer and distance-dependent spin coupling in atomically precise AgCu nanoclusters},
author = {Nan Xia and Jianpei Xing and Di Peng and Shiyu Ji and Jun Zha and Nan Yan and Yan Su and Xue Jiang and Zhi Zeng and Jijun Zhao and Zhikun Wu
},
url = {https://www.nature.com/articles/s41467-022-33651-9.pdf},
doi = {10.1038/s41467-022-33651-9},
year = {2022},
date = {2022-10-08},
urldate = {2022-10-08},
journal = {Nature Communications},
volume = {13},
number = {5934},
abstract = {Nanoparticle assembly paves the way for unanticipated properties and applications from the nanoscale to the macroscopic world. However, the study of such material systems is greatly inhibited due to the obscure compositions and structures of nanoparticles (especially the surface structures). The assembly of atomically precise nanoparticles is challenging, and such an assembly of nanoparticles with metal core sizes strictly larger than 1 nm has not been achieved yet. Here, we introduced an on-site synthesis-and-assembly strategy, and successfully obtained a straight-chain assembly structure consisting of Ag77Cu22(CHT)48 (CHT: cyclohexanethiolate) nanoparticles with two nanoparticles separated by one S atom, as revealed by mass spectrometry and single crystal X-ray crystallography. Although Ag77Cu22(CHT)48 bears one unpaired shell-closing electron, the magnetic moment is found to be mainly localized at the S linker with magnetic isotropy, and the sulfur radicals were experimentally verified and found to be unstable after disassembly, demonstrating assembly-induced spin transfer. Besides, spin nanoparticles are found to couple and lose their paramagnetism at sufficiently short inter-nanoparticle distance, namely, the spin coupling depends on the inter-nanoparticle distance. However, it is not found that the spin coupling leads to the nanoparticle growth.},
note = {The assembly of atomically precise clusters into ordered superstructures enables new functional material designs. Here, the authors propose a strategy for linear arrangements of AgCu clusters and explore the consequent transfer and coupling of magnetic spins.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanoparticle assembly paves the way for unanticipated properties and applications from the nanoscale to the macroscopic world. However, the study of such material systems is greatly inhibited due to the obscure compositions and structures of nanoparticles (especially the surface structures). The assembly of atomically precise nanoparticles is challenging, and such an assembly of nanoparticles with metal core sizes strictly larger than 1 nm has not been achieved yet. Here, we introduced an on-site synthesis-and-assembly strategy, and successfully obtained a straight-chain assembly structure consisting of Ag77Cu22(CHT)48 (CHT: cyclohexanethiolate) nanoparticles with two nanoparticles separated by one S atom, as revealed by mass spectrometry and single crystal X-ray crystallography. Although Ag77Cu22(CHT)48 bears one unpaired shell-closing electron, the magnetic moment is found to be mainly localized at the S linker with magnetic isotropy, and the sulfur radicals were experimentally verified and found to be unstable after disassembly, demonstrating assembly-induced spin transfer. Besides, spin nanoparticles are found to couple and lose their paramagnetism at sufficiently short inter-nanoparticle distance, namely, the spin coupling depends on the inter-nanoparticle distance. However, it is not found that the spin coupling leads to the nanoparticle growth. |
| 13. | 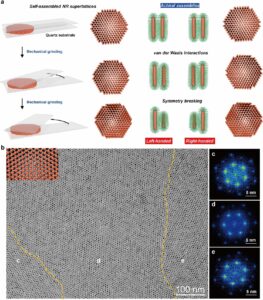 | Zhiwei Yang, Yanze Wei, Jingjing Wei, Zhijie Yang Chiral superstructures of inorganic nanorods by macroscopic mechanical grinding In: Nature Communications, vol. 13, no. 5844, 2022, (Chiroptic materials made of self-assembled nanomaterials are essential for advanced optical applications. Here, the authors show that macroscopic grinding can break the symmetry in achiral superlattices of inorganic nanorods, generating chiral superstructures.). @article{nokey,
title = {Chiral superstructures of inorganic nanorods by macroscopic mechanical grinding},
author = {Zhiwei Yang and Yanze Wei and Jingjing Wei and Zhijie Yang },
url = {https://www.nature.com/articles/s41467-022-33638-6.pdf},
doi = {10.1038/s41467-022-33638-6},
year = {2022},
date = {2022-10-04},
urldate = {2022-10-04},
journal = {Nature Communications},
volume = {13},
number = {5844},
abstract = {The development of mechanochemistry substantially expands the traditional synthetic realm at the molecular level. Here, we extend the concept of mechanochemistry from atomic/molecular solids to the nanoparticle solids, and show how the macroscopic grinding is being capable of generating chirality in self-assembled nanorod (NR) assemblies. Specifically, the weak van der Waals interaction is dominated in self-assembled NR assemblies when their surface is coated with aliphatic chains, which can be overwhelmed by a press-and-rotate mechanic force macroscopically. The chiral sign of the NR assemblies can be well-controlled by the rotating directions, where the clockwise and counter-clockwise rotation leads to the positive and negative Cotton effect in circular dichroism and circularly polarized luminescence spectra, respectively. Importantly, we show that the present approach can be applied to NRs of diverse inorganic materials, including CdSe, CdSe/CdS, and TiO2. Equally important, the as-prepared chiral NR assemblies could be served as porous yet robust chiral substrates, which enable to host other molecular materials and induce the chirality transfer from substrate to the molecular system.},
note = {Chiroptic materials made of self-assembled nanomaterials are essential for advanced optical applications. Here, the authors show that macroscopic grinding can break the symmetry in achiral superlattices of inorganic nanorods, generating chiral superstructures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The development of mechanochemistry substantially expands the traditional synthetic realm at the molecular level. Here, we extend the concept of mechanochemistry from atomic/molecular solids to the nanoparticle solids, and show how the macroscopic grinding is being capable of generating chirality in self-assembled nanorod (NR) assemblies. Specifically, the weak van der Waals interaction is dominated in self-assembled NR assemblies when their surface is coated with aliphatic chains, which can be overwhelmed by a press-and-rotate mechanic force macroscopically. The chiral sign of the NR assemblies can be well-controlled by the rotating directions, where the clockwise and counter-clockwise rotation leads to the positive and negative Cotton effect in circular dichroism and circularly polarized luminescence spectra, respectively. Importantly, we show that the present approach can be applied to NRs of diverse inorganic materials, including CdSe, CdSe/CdS, and TiO2. Equally important, the as-prepared chiral NR assemblies could be served as porous yet robust chiral substrates, which enable to host other molecular materials and induce the chirality transfer from substrate to the molecular system. |
| 12. | 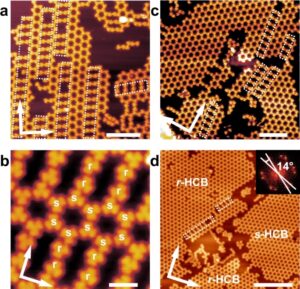 | Zhen-Yu Yi, Xue-Qing Yang, Jun-Jie Duan, Xiong Zhou, Ting Chen, Dong Wang, Li-Jun Wan Evolution of Br⋯Br contacts in enantioselective molecular recognition during chiral 2D crystallization In: Nature Communications, vol. 13, no. 5850, 2022, (Halogen-mediated interactions control molecular recognition in many chemical and biological systems. Here, the authors demonstrate two types of Br⋯Br contacts and their importance in chiral on-surface crystallization.). @article{nokey,
title = {Evolution of Br⋯Br contacts in enantioselective molecular recognition during chiral 2D crystallization},
author = {Zhen-Yu Yi and Xue-Qing Yang and Jun-Jie Duan and Xiong Zhou and Ting Chen and Dong Wang and Li-Jun Wan },
url = {https://www.nature.com/articles/s41467-022-33446-y.pdf},
doi = {10.1038/s41467-022-33446-y},
year = {2022},
date = {2022-10-04},
urldate = {2022-10-04},
journal = {Nature Communications},
volume = {13},
number = {5850},
abstract = {Halogen-mediated interactions play an important role in molecular recognition and crystallization in many chemical and biological systems, whereas their effect on homochiral versus heterochiral recognition and crystallization has rarely been explored. Here we demonstrate the evolution of Br⋯Br contacts in chiral recognition during 2D crystallization. On Ag(100), type I contacts prevail at low coverage and lead to homochiral recognition and the formation of 2D conglomerates; whereas type II contacts mediating heterochiral recognition are suppressed at medium coverage and appear in the racemates induced by structural transitions at high coverage. On Ag(111), type I contacts dominate the 2D crystallization and generate 2D conglomerates exclusively. DFT calculations suggest that the energy difference between type I and type II contacts is reversed upon adsorption due to the substrate induced mismatch energy penalty. This result provides fundamental understanding of halogen-mediated interactions in molecular recognition and crystallization on surface.},
note = {Halogen-mediated interactions control molecular recognition in many chemical and biological systems. Here, the authors demonstrate two types of Br⋯Br contacts and their importance in chiral on-surface crystallization.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Halogen-mediated interactions play an important role in molecular recognition and crystallization in many chemical and biological systems, whereas their effect on homochiral versus heterochiral recognition and crystallization has rarely been explored. Here we demonstrate the evolution of Br⋯Br contacts in chiral recognition during 2D crystallization. On Ag(100), type I contacts prevail at low coverage and lead to homochiral recognition and the formation of 2D conglomerates; whereas type II contacts mediating heterochiral recognition are suppressed at medium coverage and appear in the racemates induced by structural transitions at high coverage. On Ag(111), type I contacts dominate the 2D crystallization and generate 2D conglomerates exclusively. DFT calculations suggest that the energy difference between type I and type II contacts is reversed upon adsorption due to the substrate induced mismatch energy penalty. This result provides fundamental understanding of halogen-mediated interactions in molecular recognition and crystallization on surface. |
| 11. | 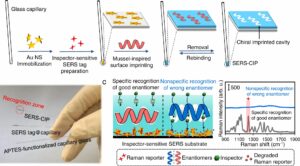 | Maryam Arabi, Abbas Ostovan, Yunqing Wang, Rongchao Mei, Longwen Fu, Jinhua Li, Xiaoyan Wang, Lingxin Chen Chiral molecular imprinting-based SERS detection strategy for absolute enantiomeric discrimination In: Nature Communications, vol. 13, no. 5757, 2022, (Absolute chiral discrimination in chiral imprinted systems is complicated by the nonspecific binding of enantiomers. Here, the authors report a SERS “inspector” recognition mechanism to distinguish between specifically and nonspecifically bound enantiomers, even in seawater and urine.). @article{nokey,
title = {Chiral molecular imprinting-based SERS detection strategy for absolute enantiomeric discrimination},
author = {Maryam Arabi and Abbas Ostovan and Yunqing Wang and Rongchao Mei and Longwen Fu and Jinhua Li and Xiaoyan Wang and Lingxin Chen},
url = {https://www.nature.com/articles/s41467-022-33448-w.pdf},
doi = {10.1038/s41467-022-33448-w},
year = {2022},
date = {2022-10-01},
urldate = {2022-10-01},
journal = {Nature Communications},
volume = {13},
number = {5757},
abstract = {Chiral discrimination is critical in environmental and life sciences. However, an ideal chiral discrimination strategy has not yet been developed because of the inevitable nonspecific binding entity of wrong enantiomers or insufficient intrinsic optical activities of chiral molecules. Here, we propose an “inspector” recognition mechanism (IRM), which is implemented on a chiral imprinted polydopamine (PDA) layer coated on surface-enhanced Raman scattering (SERS) tag layer. The IRM works based on the permeability change of the imprinted PDA after the chiral recognition and scrutiny of the permeability by an inspector molecule. Good enantiomer can specifically recognize and fully fill the chiral imprinted cavities, whereas the wrong cannot. Then a linear shape aminothiol molecule, as an inspector of the recognition status is introduced, which can only percolate through the vacant and nonspecifically occupied cavities, inducing the SERS signal to decrease. Accordingly, chirality information exclusively stems from good enantiomer specific binding, while nonspecific recognition of wrong enantiomer is curbed. The IRM benefits from sensitivity and versatility, enabling absolute discrimination of a wide variety of chiral molecules regardless of size, functional groups, polarities, optical activities, Raman scattering, and the number of chiral centers.},
note = {Absolute chiral discrimination in chiral imprinted systems is complicated by the nonspecific binding of enantiomers. Here, the authors report a SERS “inspector” recognition mechanism to distinguish between specifically and nonspecifically bound enantiomers, even in seawater and urine.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chiral discrimination is critical in environmental and life sciences. However, an ideal chiral discrimination strategy has not yet been developed because of the inevitable nonspecific binding entity of wrong enantiomers or insufficient intrinsic optical activities of chiral molecules. Here, we propose an “inspector” recognition mechanism (IRM), which is implemented on a chiral imprinted polydopamine (PDA) layer coated on surface-enhanced Raman scattering (SERS) tag layer. The IRM works based on the permeability change of the imprinted PDA after the chiral recognition and scrutiny of the permeability by an inspector molecule. Good enantiomer can specifically recognize and fully fill the chiral imprinted cavities, whereas the wrong cannot. Then a linear shape aminothiol molecule, as an inspector of the recognition status is introduced, which can only percolate through the vacant and nonspecifically occupied cavities, inducing the SERS signal to decrease. Accordingly, chirality information exclusively stems from good enantiomer specific binding, while nonspecific recognition of wrong enantiomer is curbed. The IRM benefits from sensitivity and versatility, enabling absolute discrimination of a wide variety of chiral molecules regardless of size, functional groups, polarities, optical activities, Raman scattering, and the number of chiral centers. |
| 10. |  | Lei Lei, Yubin Wang, Weixin Xu, Renguang Ye, Youjie Hua, Degang Deng, Liang Chen, Paras N. Prasad, Shiqing Xu Manipulation of time-dependent multicolour evolution of X-ray excited afterglow in lanthanide-doped fluoride nanoparticles In: Nature Communications, vol. 13, no. 5739, 2022, (X-ray activated afterglow nanomaterials are desirable components for advanced optoelectronic applications. Here, the authors present pathways to modulate the stimulus-responsive color emissions in lanthanide-doped fluoride core-shell nanoparticles.). @article{nokey,
title = {Manipulation of time-dependent multicolour evolution of X-ray excited afterglow in lanthanide-doped fluoride nanoparticles},
author = {Lei Lei and Yubin Wang and Weixin Xu and Renguang Ye and Youjie Hua and Degang Deng and Liang Chen and Paras N. Prasad and Shiqing Xu },
editor = {Felix Allum},
url = {https://www.nature.com/articles/s41467-022-33489-1.pdf},
doi = {10.1038/s41467-022-33489-1},
year = {2022},
date = {2022-09-30},
urldate = {2022-09-30},
journal = {Nature Communications},
volume = {13},
number = {5739},
abstract = {External manipulation of emission colour is of significance for scientific research and applications, however, the general stimulus-responsive colour modulation method requires both stringent control of microstructures and continously adjustment of particular stimuli conditions. Here, we introduce pathways to manipulate the kinetics of time evolution of both intensity and spectral characteristics of X-ray excited afterglow (XEA) by regioselective doping of lanthanide activators in core-shell nanostructures. Our work reported here reveals the following phenomena: 1. The XEA intensities of multiple lanthanide activators are significantly enhanced via incorporating interstitial Na+ ions inside the nanocrystal structure. 2. The XEA intensities of activators exhibit diverse decay rates in the core and the shell and can largely be tuned separately, which enables us to realize a series of core@shell NPs featuring distinct time-dependent afterglow colour evolution. 3. A core/multi-shell NP structure can be designed to simultaneously generate afterglow, upconversion and downshifting to realize multimode time-dependent multicolour evolutions. These findings can promote the development of superior XEA and plentiful spectral manipulation, opening up a broad range of applications ranging from multiplexed biosensing, to high-capacity information encryption, to multidimensional displays and to multifunctional optoelectronic devices.},
note = {X-ray activated afterglow nanomaterials are desirable components for advanced optoelectronic applications. Here, the authors present pathways to modulate the stimulus-responsive color emissions in lanthanide-doped fluoride core-shell nanoparticles.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
External manipulation of emission colour is of significance for scientific research and applications, however, the general stimulus-responsive colour modulation method requires both stringent control of microstructures and continously adjustment of particular stimuli conditions. Here, we introduce pathways to manipulate the kinetics of time evolution of both intensity and spectral characteristics of X-ray excited afterglow (XEA) by regioselective doping of lanthanide activators in core-shell nanostructures. Our work reported here reveals the following phenomena: 1. The XEA intensities of multiple lanthanide activators are significantly enhanced via incorporating interstitial Na+ ions inside the nanocrystal structure. 2. The XEA intensities of activators exhibit diverse decay rates in the core and the shell and can largely be tuned separately, which enables us to realize a series of core@shell NPs featuring distinct time-dependent afterglow colour evolution. 3. A core/multi-shell NP structure can be designed to simultaneously generate afterglow, upconversion and downshifting to realize multimode time-dependent multicolour evolutions. These findings can promote the development of superior XEA and plentiful spectral manipulation, opening up a broad range of applications ranging from multiplexed biosensing, to high-capacity information encryption, to multidimensional displays and to multifunctional optoelectronic devices. |
| 9. |  | Jing Ai, Xueliang Zhang, Te Bai, Qing Shen, Peter Oleynikov, Yingying Duan, Osamu Terasaki, Shunai Che, Lu Han Synchronous quantitative analysis of chiral mesostructured inorganic crystals by 3D electron diffraction tomography In: Nature Communications, vol. 13, no. 5718, 2022, (Chiral mesostructured inorganic crystals exhibit distinctive twisting and helical hierarchical stacking. Here, the authors report a general approach for the synchronous quantitative analysis of rotation axis, torsion angle, pitch length, and arrangement modes.). @article{nokey,
title = {Synchronous quantitative analysis of chiral mesostructured inorganic crystals by 3D electron diffraction tomography},
author = {Jing Ai and Xueliang Zhang and Te Bai and Qing Shen and Peter Oleynikov and Yingying Duan and Osamu Terasaki and Shunai Che and Lu Han},
url = {https://www.nature.com/articles/s41467-022-33443-1.pdf},
doi = {10.1038/s41467-022-33443-1},
year = {2022},
date = {2022-09-29},
urldate = {2022-09-29},
journal = {Nature Communications},
volume = {13},
number = {5718},
abstract = {Chiral mesostructures exhibit distinctive twisting and helical hierarchical stacking ranging from atomic to micrometre scales with fascinating structural-chiral anisotropy properties. However, the detailed determination of their multilevel chirality remains challenging due to the limited information from spectroscopy, diffraction techniques, scanning electron microscopy and the two-dimensional projections in transmission electron microscopy. Herein, we report a general approach to determine chiral hierarchical mesostructures based on three-dimensional electron diffraction tomography (3D EDT), by which the structure can be solved synchronously according to the quantitative measurement of diffraction spot deformations and their arrangement in reciprocal space. This method was verified on two samples—chiral mesostructured nickel molybdate and chiral mesostructured tin dioxide—revealing hierarchical chiral structures that cannot be determined by conventional techniques. This approach provides more precise and comprehensive identification of the hierarchical mesostructures, which is expected to advance our understanding of structural–chiral anisotropy at the fundamental level.
},
note = {Chiral mesostructured inorganic crystals exhibit distinctive twisting and helical hierarchical stacking. Here, the authors report a general approach for the synchronous quantitative analysis of rotation axis, torsion angle, pitch length, and arrangement modes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chiral mesostructures exhibit distinctive twisting and helical hierarchical stacking ranging from atomic to micrometre scales with fascinating structural-chiral anisotropy properties. However, the detailed determination of their multilevel chirality remains challenging due to the limited information from spectroscopy, diffraction techniques, scanning electron microscopy and the two-dimensional projections in transmission electron microscopy. Herein, we report a general approach to determine chiral hierarchical mesostructures based on three-dimensional electron diffraction tomography (3D EDT), by which the structure can be solved synchronously according to the quantitative measurement of diffraction spot deformations and their arrangement in reciprocal space. This method was verified on two samples—chiral mesostructured nickel molybdate and chiral mesostructured tin dioxide—revealing hierarchical chiral structures that cannot be determined by conventional techniques. This approach provides more precise and comprehensive identification of the hierarchical mesostructures, which is expected to advance our understanding of structural–chiral anisotropy at the fundamental level.
|
| 8. |  | Corentin Dabard, Victor Guilloux, Charlie Gréboval, Hong Po, Lina Makke, Ningyuan Fu, Xiang Zhen Xu, Mathieu G. Silly, Gilles Patriarche, Emmanuel Lhuillier, Thierry Barisien, Juan I. Climente, Benjamin T. Diroll, Sandrine Ithurria Double-crowned 2D semiconductor nanoplatelets with bicolor power-tunable emission In: Nature Communications, vol. 13, no. 5094, 2022, (Nanocrystals are desirable light sources for advanced display technologies. Here, the authors report on double-crowned 2D semiconductor nanoplatelets as light downconverters that offer both green and red emissions to achieve a wide color gamut.). @article{nokey,
title = {Double-crowned 2D semiconductor nanoplatelets with bicolor power-tunable emission},
author = {Corentin Dabard and Victor Guilloux and Charlie Gréboval and Hong Po and Lina Makke and Ningyuan Fu and Xiang Zhen Xu and Mathieu G. Silly and Gilles Patriarche and Emmanuel Lhuillier and Thierry Barisien and Juan I. Climente and Benjamin T. Diroll and Sandrine Ithurria},
url = {https://www.nature.com/articles/s41467-022-32713-2.pdf},
doi = {10.1038/s41467-022-32713-2},
year = {2022},
date = {2022-08-30},
urldate = {2022-08-30},
journal = {Nature Communications},
volume = {13},
number = {5094},
abstract = {Nanocrystals (NCs) are now established building blocks for optoelectronics and their use as down converters for large gamut displays has been their first mass market. NC integration relies on a combination of green and red NCs into a blend, which rises post-growth formulation issues. A careful engineering of the NCs may enable dual emissions from a single NC population which violates Kasha’s rule, which stipulates that emission should occur at the band edge. Thus, in addition to an attentive control of band alignment to obtain green and red signals, non-radiative decay paths also have to be carefully slowed down to enable emission away from the ground state. Here, we demonstrate that core/crown/crown 2D nanoplatelets (NPLs), made of CdSe/CdTe/CdSe, can combine a large volume and a type-II band alignment enabling simultaneously red and narrow green emissions. Moreover, we demonstrate that the ratio of the two emissions can be tuned by the incident power, which results in a saturation of the red emission due to non-radiative Auger recombination that affects this emission much stronger than the green one. Finally, we also show that dual-color, power tunable, emission can be obtained through an electrical excitation.},
note = {Nanocrystals are desirable light sources for advanced display technologies. Here, the authors report on double-crowned 2D semiconductor nanoplatelets as light downconverters that offer both green and red emissions to achieve a wide color gamut.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nanocrystals (NCs) are now established building blocks for optoelectronics and their use as down converters for large gamut displays has been their first mass market. NC integration relies on a combination of green and red NCs into a blend, which rises post-growth formulation issues. A careful engineering of the NCs may enable dual emissions from a single NC population which violates Kasha’s rule, which stipulates that emission should occur at the band edge. Thus, in addition to an attentive control of band alignment to obtain green and red signals, non-radiative decay paths also have to be carefully slowed down to enable emission away from the ground state. Here, we demonstrate that core/crown/crown 2D nanoplatelets (NPLs), made of CdSe/CdTe/CdSe, can combine a large volume and a type-II band alignment enabling simultaneously red and narrow green emissions. Moreover, we demonstrate that the ratio of the two emissions can be tuned by the incident power, which results in a saturation of the red emission due to non-radiative Auger recombination that affects this emission much stronger than the green one. Finally, we also show that dual-color, power tunable, emission can be obtained through an electrical excitation. |
| 7. |  | Lukas Grote, Martin Seyrich, Ralph Döhrmann, Sani Harouna-Mayer, Federica Mancini, Emilis Kaziukenas, Irene Fernandez-Cuesta, Cecilia Zito, Olga Vasylieva, Felix Wittwer, Michal Odstrcil, Natnael Mogos, Mirko Landmann, Christian Schroer, Dorota Koziej Imaging Cu2O nanocube hollowing in solution by quantitative in-situ X-ray ptychography In: Nature Communications, vol. 13, no. 4971, 2022, (Observing morphological changes of nanoparticles in solution requires advanced in-situ imaging methods. Here, the authors use X-ray ptychography to image the growth and hollowing of Cu2O nanocubes in 3D.). @article{nokey,
title = {Imaging Cu2O nanocube hollowing in solution by quantitative in-situ X-ray ptychography},
author = {Lukas Grote and Martin Seyrich and Ralph Döhrmann and Sani Harouna-Mayer and Federica Mancini and Emilis Kaziukenas and Irene Fernandez-Cuesta and Cecilia Zito and Olga Vasylieva and Felix Wittwer and Michal Odstrcil and Natnael Mogos and Mirko Landmann and Christian Schroer and Dorota Koziej},
url = {https://www.nature.com/articles/s41467-022-32373-2.pdf},
doi = {10.1038/s41467-022-32373-2},
year = {2022},
date = {2022-08-29},
urldate = {2022-08-29},
journal = {Nature Communications},
volume = {13},
number = {4971},
abstract = {Understanding morphological changes of nanoparticles in solution is essential to tailor the functionality of devices used in energy generation and storage. However, we lack experimental methods that can visualize these processes in solution, or in electrolyte, and provide three-dimensional information. Here, we show how X-ray ptychography enables in situ nano-imaging of the formation and hollowing of nanoparticles in solution at 155°C. We simultaneously image the growth of about 100 nanocubes with a spatial resolution of 66 nm. The quantitative phase images give access to the third dimension, allowing to additionally study particle thickness. We reveal that the substrate hinders their out-of-plane growth, thus the nanocubes are in fact nanocuboids. Moreover, we observe that the reduction of Cu2O to Cu triggers the hollowing of the nanocuboids. We critically assess the interaction of X-rays with the liquid sample. Our method enables detailed in-solution imaging for a wide range of reaction conditions.},
note = {Observing morphological changes of nanoparticles in solution requires advanced in-situ imaging methods. Here, the authors use X-ray ptychography to image the growth and hollowing of Cu2O nanocubes in 3D.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Understanding morphological changes of nanoparticles in solution is essential to tailor the functionality of devices used in energy generation and storage. However, we lack experimental methods that can visualize these processes in solution, or in electrolyte, and provide three-dimensional information. Here, we show how X-ray ptychography enables in situ nano-imaging of the formation and hollowing of nanoparticles in solution at 155°C. We simultaneously image the growth of about 100 nanocubes with a spatial resolution of 66 nm. The quantitative phase images give access to the third dimension, allowing to additionally study particle thickness. We reveal that the substrate hinders their out-of-plane growth, thus the nanocubes are in fact nanocuboids. Moreover, we observe that the reduction of Cu2O to Cu triggers the hollowing of the nanocuboids. We critically assess the interaction of X-rays with the liquid sample. Our method enables detailed in-solution imaging for a wide range of reaction conditions. |
| 6. |  | Hajir Hilal, Qiang Zhao, Jeongwon Kim, Sungwoo Lee, MohammadNavid Haddadnezhad, Sungjae Yoo, Soohyun Lee, Woongkyu Park, Woocheol Park, Jaewon Lee, Joong Wook Lee, Insub Jung, Sungho Park Three-dimensional nanoframes with dual rims as nanoprobes for biosensing In: Nature Communications, vol. 13, no. 4813, 2022, (Most SERS-active nanostructures suffer from low robustness against misalignment to field polarization. Here, the authors demonstrate three-dimensional nanoframes of octahedral geometry, with two rims engraved on each facet, as polarization-independent SERS nanoprobes.). @article{nokey,
title = {Three-dimensional nanoframes with dual rims as nanoprobes for biosensing},
author = {Hajir Hilal and Qiang Zhao and Jeongwon Kim and Sungwoo Lee and MohammadNavid Haddadnezhad and Sungjae Yoo and Soohyun Lee and Woongkyu Park and Woocheol Park and Jaewon Lee and Joong Wook Lee and Insub Jung and Sungho Park },
url = {https://www.nature.com/articles/s41467-022-32549-w.pdf},
doi = {10.1038/s41467-022-32549-w},
year = {2022},
date = {2022-08-16},
urldate = {2022-08-16},
journal = {Nature Communications},
volume = {13},
number = {4813},
abstract = {Three-dimensional (3D) nanoframe structures are very appealing because their inner voids and ridges interact efficiently with light and analytes, allowing for effective optical-based sensing. However, the realization of complex nanoframe architecture with high yield is challenging because the systematic design of such a complicated nanostructure lacks an appropriate synthesis protocol. Here, we show the synthesis method for complex 3D nanoframes wherein two-dimensional (2D) dual-rim nanostructures are engraved on each facet of octahedral nanoframes. The synthetic scheme proceeds through multiple executable on-demand steps. With Au octahedral nanoparticles as a sacrificial template, sequential processes of edge-selective Pt deposition and inner Au etching lead to Pt octahedral mono-rim nanoframes. Then, adlayers of Au are grown on Pt skeletons via the Frank-van der Merwe mode, forming sharp and well-developed edges. Next, Pt selective deposition on both the inner and outer boundaries leads to tunable geometric patterning on Au. Finally, after the selective etching of Au, Pt octahedral dual-rim nanoframes with highly homogeneous size and shape are achieved. In order to endow plasmonic features, Au is coated around Pt frames while retaining their geometric shape. The resultant plasmonic dual-rim engraved nanoframes possess strong light entrapping capability verified by single-particle surface-enhanced Raman scattering (SERS) and show the potential of nanoprobes for biosensing through SERS-based immunoassay.},
note = {Most SERS-active nanostructures suffer from low robustness against misalignment to field polarization. Here, the authors demonstrate three-dimensional nanoframes of octahedral geometry, with two rims engraved on each facet, as polarization-independent SERS nanoprobes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Three-dimensional (3D) nanoframe structures are very appealing because their inner voids and ridges interact efficiently with light and analytes, allowing for effective optical-based sensing. However, the realization of complex nanoframe architecture with high yield is challenging because the systematic design of such a complicated nanostructure lacks an appropriate synthesis protocol. Here, we show the synthesis method for complex 3D nanoframes wherein two-dimensional (2D) dual-rim nanostructures are engraved on each facet of octahedral nanoframes. The synthetic scheme proceeds through multiple executable on-demand steps. With Au octahedral nanoparticles as a sacrificial template, sequential processes of edge-selective Pt deposition and inner Au etching lead to Pt octahedral mono-rim nanoframes. Then, adlayers of Au are grown on Pt skeletons via the Frank-van der Merwe mode, forming sharp and well-developed edges. Next, Pt selective deposition on both the inner and outer boundaries leads to tunable geometric patterning on Au. Finally, after the selective etching of Au, Pt octahedral dual-rim nanoframes with highly homogeneous size and shape are achieved. In order to endow plasmonic features, Au is coated around Pt frames while retaining their geometric shape. The resultant plasmonic dual-rim engraved nanoframes possess strong light entrapping capability verified by single-particle surface-enhanced Raman scattering (SERS) and show the potential of nanoprobes for biosensing through SERS-based immunoassay. |
| 5. |  | Lei Zhang, Chen Yang, Chenxi Lu, Xingxing Li, Yilin Guo, Jianning Zhang, Jinglong Lin, Zhizhou Li, Chuancheng Jia, Jinlong Yang, K. N. Houk, Fanyang Mo, Xuefeng Guo Precise electrical gating of the single-molecule Mizoroki-Heck reaction In: Nature Communications, vol. 13, no. 4552, 2022, (Guiding chemical reactions in a predictable and controllable manner is an ultimate goal of chemistry. Here, the authors show tuning of the single-molecule Mizoroki-Heck catalytic cycle through electrical gating and direct in-situ detection.). @article{nokey,
title = {Precise electrical gating of the single-molecule Mizoroki-Heck reaction},
author = {Lei Zhang and Chen Yang and Chenxi Lu and Xingxing Li and Yilin Guo and Jianning Zhang and Jinglong Lin and Zhizhou Li and Chuancheng Jia and Jinlong Yang and K. N. Houk and Fanyang Mo and Xuefeng Guo },
url = {https://www.nature.com/articles/s41467-022-32351-8.pdf},
doi = {10.1038/s41467-022-32351-8},
year = {2022},
date = {2022-08-05},
urldate = {2022-08-05},
journal = {Nature Communications},
volume = {13},
number = {4552},
abstract = {Precise tuning of chemical reactions with predictable and controllable manners, an ultimate goal chemists desire to achieve, is valuable in the scientific community. This tunability is necessary to understand and regulate chemical transformations at both macroscopic and single-molecule levels to meet demands in potential application scenarios. Herein, we realise accurate tuning of a single-molecule Mizoroki-Heck reaction via applying gate voltages as well as complete deciphering of its detailed intrinsic mechanism by employing an in-situ electrical single-molecule detection, which possesses the capability of single-event tracking. The Mizoroki-Heck reaction can be regulated in different dimensions with a constant catalyst molecule, including the molecular orbital gating of Pd(0) catalyst, the on/off switching of the Mizoroki-Heck reaction, the promotion of its turnover frequency, and the regulation of each elementary reaction within the Mizoroki-Heck catalytic cycle. These results extend the tuning scope of chemical reactions from the macroscopic view to the single-molecule approach, inspiring new insights into designing different strategies or devices to unveil reaction mechanisms and discover novel phenomena.},
note = {Guiding chemical reactions in a predictable and controllable manner is an ultimate goal of chemistry. Here, the authors show tuning of the single-molecule Mizoroki-Heck catalytic cycle through electrical gating and direct in-situ detection.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Precise tuning of chemical reactions with predictable and controllable manners, an ultimate goal chemists desire to achieve, is valuable in the scientific community. This tunability is necessary to understand and regulate chemical transformations at both macroscopic and single-molecule levels to meet demands in potential application scenarios. Herein, we realise accurate tuning of a single-molecule Mizoroki-Heck reaction via applying gate voltages as well as complete deciphering of its detailed intrinsic mechanism by employing an in-situ electrical single-molecule detection, which possesses the capability of single-event tracking. The Mizoroki-Heck reaction can be regulated in different dimensions with a constant catalyst molecule, including the molecular orbital gating of Pd(0) catalyst, the on/off switching of the Mizoroki-Heck reaction, the promotion of its turnover frequency, and the regulation of each elementary reaction within the Mizoroki-Heck catalytic cycle. These results extend the tuning scope of chemical reactions from the macroscopic view to the single-molecule approach, inspiring new insights into designing different strategies or devices to unveil reaction mechanisms and discover novel phenomena. |
| 4. | 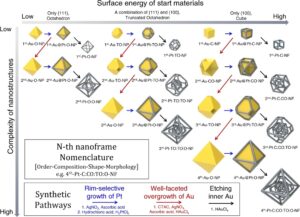 | Sungjae Yoo, Jaewon Lee, Hajir Hilal, Insub Jung, Woongkyu Park, Joong Wook Lee, Soobong Choi, Sungho Park Nesting of multiple polyhedral plasmonic nanoframes into a single entity In: Nature Communications, vol. 13, no. 4544, 2022, (The spatial configuration of nanostructure building blocks determines the physical and optical properties of their superstructures. Here, the authors report on complex nanoparticles in which different geometric forms of nanoframes are nested into a single entity by multistep chemical reactions.). @article{nokey,
title = {Nesting of multiple polyhedral plasmonic nanoframes into a single entity},
author = {Sungjae Yoo and Jaewon Lee and Hajir Hilal and Insub Jung and Woongkyu Park and Joong Wook Lee and Soobong Choi and Sungho Park},
url = {https://www.nature.com/articles/s41467-022-32261-9.pdf},
doi = {10.1038/s41467-022-32261-9},
year = {2022},
date = {2022-08-04},
urldate = {2022-08-04},
journal = {Nature Communications},
volume = {13},
number = {4544},
abstract = {The development of plasmonic nanostructures with intricate nanoframe morphologies has attracted considerable interest for improving catalytic and optical properties. However, arranging multiple nanoframes in one nanostructure especially, in a solution phase remains a great challenge. Herein, we show complex nanoparticles by embedding various shapes of three-dimensional polyhedral nanoframes within a single entity through rationally designed synthetic pathways. This synthetic strategy is based on the selective deposition of platinum atoms on high surface energy facets and subsequent growth into solid platonic nanoparticles, followed by the etching of inner Au domains, leaving complex nanoframes. Our synthetic routes are rationally designed and executable on-demand with a high structural controllability. Diverse Au solid nanostructures (octahedra, truncated octahedra, cuboctahedra, and cubes) evolved into complex multi-layered nanoframes with different numbers/shapes/sizes of internal nanoframes. After coating the surface of the nanoframes with plasmonically active metal (like Ag), the materials exhibited highly enhanced electromagnetic near-field focusing embedded within the internal complicated rim architecture.},
note = {The spatial configuration of nanostructure building blocks determines the physical and optical properties of their superstructures. Here, the authors report on complex nanoparticles in which different geometric forms of nanoframes are nested into a single entity by multistep chemical reactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The development of plasmonic nanostructures with intricate nanoframe morphologies has attracted considerable interest for improving catalytic and optical properties. However, arranging multiple nanoframes in one nanostructure especially, in a solution phase remains a great challenge. Herein, we show complex nanoparticles by embedding various shapes of three-dimensional polyhedral nanoframes within a single entity through rationally designed synthetic pathways. This synthetic strategy is based on the selective deposition of platinum atoms on high surface energy facets and subsequent growth into solid platonic nanoparticles, followed by the etching of inner Au domains, leaving complex nanoframes. Our synthetic routes are rationally designed and executable on-demand with a high structural controllability. Diverse Au solid nanostructures (octahedra, truncated octahedra, cuboctahedra, and cubes) evolved into complex multi-layered nanoframes with different numbers/shapes/sizes of internal nanoframes. After coating the surface of the nanoframes with plasmonically active metal (like Ag), the materials exhibited highly enhanced electromagnetic near-field focusing embedded within the internal complicated rim architecture. |
| 3. |  | Mingu Kang, Hyun Woo Kim, Elham Oleiki, Yeonjeong Koo, Hyeongwoo Lee, Huitae Joo, Jinseong Choi, Taeyong Eom, Geunsik Lee, Yung Doug Suh, Kyoung-Duck Park Conformational heterogeneity of molecules physisorbed on a gold surface at room temperature In: Nature Communications, vol. 13, no. 4133, 2022, (Tip-enhanced vibrational spectroscopy at room temperature is complicated by molecular conformational dynamics, photobleaching, contaminations, and chemical reactions in air. This study demonstrates that a sub-nm protective layer of Al2O3 provides robust conditions for probing single-molecule conformations.). @article{Kang2022,
title = {Conformational heterogeneity of molecules physisorbed on a gold surface at room temperature},
author = {Mingu Kang and Hyun Woo Kim and Elham Oleiki and Yeonjeong Koo and Hyeongwoo Lee and Huitae Joo and Jinseong Choi and Taeyong Eom and Geunsik Lee and Yung Doug Suh and Kyoung-Duck Park},
url = {https://www.nature.com/articles/s41467-022-31576-x.pdf},
doi = {10.1038/s41467-022-31576-x},
year = {2022},
date = {2022-07-15},
urldate = {2022-07-15},
journal = {Nature Communications},
volume = {13},
number = {4133},
abstract = {A quantitative single-molecule tip-enhanced Raman spectroscopy (TERS) study at room temperature remained a challenge due to the rapid structural dynamics of molecules exposed to air. Here, we demonstrate the hyperspectral TERS imaging of single or a few brilliant cresyl blue (BCB) molecules at room temperature, along with quantitative spectral analyses. Robust chemical imaging is enabled by the freeze-frame approach using a thin Al2O3 capping layer, which suppresses spectral diffusions and inhibits chemical reactions and contamination in air. For the molecules resolved spatially in the TERS image, a clear Raman peak variation up to 7.5 cm−1 is observed, which cannot be found in molecular ensembles. From density functional theory-based quantitative analyses of the varied TERS peaks, we reveal the conformational heterogeneity at the single-molecule level. This work provides a facile way to investigate the single-molecule properties in interacting media, expanding the scope of single-molecule vibrational spectroscopy studies.},
note = {Tip-enhanced vibrational spectroscopy at room temperature is complicated by molecular conformational dynamics, photobleaching, contaminations, and chemical reactions in air. This study demonstrates that a sub-nm protective layer of Al2O3 provides robust conditions for probing single-molecule conformations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
A quantitative single-molecule tip-enhanced Raman spectroscopy (TERS) study at room temperature remained a challenge due to the rapid structural dynamics of molecules exposed to air. Here, we demonstrate the hyperspectral TERS imaging of single or a few brilliant cresyl blue (BCB) molecules at room temperature, along with quantitative spectral analyses. Robust chemical imaging is enabled by the freeze-frame approach using a thin Al2O3 capping layer, which suppresses spectral diffusions and inhibits chemical reactions and contamination in air. For the molecules resolved spatially in the TERS image, a clear Raman peak variation up to 7.5 cm−1 is observed, which cannot be found in molecular ensembles. From density functional theory-based quantitative analyses of the varied TERS peaks, we reveal the conformational heterogeneity at the single-molecule level. This work provides a facile way to investigate the single-molecule properties in interacting media, expanding the scope of single-molecule vibrational spectroscopy studies. |
| 2. |  | Nam Heon Cho, Young Bi Kim, Yoon Young Lee, Sang Won Im, Ryeong Myeong Kim, Jeong Won Kim, Seok Daniel Namgung, Hye-Eun Lee, Hyeohn Kim, Jeong Hyun Han, Hye Won Chung, Yoon Ho Lee, Jeong Woo Han, Ki Tae Nam Adenine oligomer directed synthesis of chiral gold nanoparticles In: Nature Communications, vol. 13, no. 3831, 2022, (Chiral plasmonic nanoparticles are of great interest in nanotechnology. Here, the authors demonstrate chiral shape guidance by single-stranded oligonucleotides during particle growth based on sequence-specific hydrogen bonding within the strand.). @article{nokey,
title = {Adenine oligomer directed synthesis of chiral gold nanoparticles},
author = {Nam Heon Cho and Young Bi Kim and Yoon Young Lee and Sang Won Im and Ryeong Myeong Kim and Jeong Won Kim and Seok Daniel Namgung and Hye-Eun Lee and Hyeohn Kim and Jeong Hyun Han and Hye Won Chung and Yoon Ho Lee and Jeong Woo Han and Ki Tae Nam },
editor = {Christian Kuttner (Ed.)},
url = {https://www.nature.com/articles/s41467-022-31513-y.pdf},
doi = {10.1038/s41467-022-31513-y},
year = {2022},
date = {2022-07-02},
urldate = {2022-07-02},
journal = {Nature Communications},
volume = {13},
number = {3831},
abstract = {Precise control of morphology and optical response of 3-dimensional chiral nanoparticles remain as a significant challenge. This work demonstrates chiral gold nanoparticle synthesis using single-stranded oligonucleotide as a chiral shape modifier. The homo-oligonucleotide composed of Adenine nucleobase specifically show a distinct chirality development with a dissymmetric factor up to g ~ 0.04 at visible wavelength, whereas other nucleobases show no development of chirality. The synthesized nanoparticle shows a counter-clockwise rotation of generated chiral arms with approximately 200 nm edge length. The molecular dynamics and density functional theory simulations reveal that Adenine shows the highest enantioselective interaction with Au(321)R/S facet in terms of binding orientation and affinity. This is attributed to the formation of sequence-specific intra-strand hydrogen bonding between nucleobases. We also found that different sequence programming of Adenine-and Cytosine-based oligomers result in chiral gold nanoparticles’ morphological and optical change. These results extend our understanding of the biomolecule-directed synthesis of chiral gold nanoparticles to sequence programmable deoxyribonucleic acid and provides a foundation for programmable synthesis of chiral gold nanoparticles.},
note = {Chiral plasmonic nanoparticles are of great interest in nanotechnology. Here, the authors demonstrate chiral shape guidance by single-stranded oligonucleotides during particle growth based on sequence-specific hydrogen bonding within the strand.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Precise control of morphology and optical response of 3-dimensional chiral nanoparticles remain as a significant challenge. This work demonstrates chiral gold nanoparticle synthesis using single-stranded oligonucleotide as a chiral shape modifier. The homo-oligonucleotide composed of Adenine nucleobase specifically show a distinct chirality development with a dissymmetric factor up to g ~ 0.04 at visible wavelength, whereas other nucleobases show no development of chirality. The synthesized nanoparticle shows a counter-clockwise rotation of generated chiral arms with approximately 200 nm edge length. The molecular dynamics and density functional theory simulations reveal that Adenine shows the highest enantioselective interaction with Au(321)R/S facet in terms of binding orientation and affinity. This is attributed to the formation of sequence-specific intra-strand hydrogen bonding between nucleobases. We also found that different sequence programming of Adenine-and Cytosine-based oligomers result in chiral gold nanoparticles’ morphological and optical change. These results extend our understanding of the biomolecule-directed synthesis of chiral gold nanoparticles to sequence programmable deoxyribonucleic acid and provides a foundation for programmable synthesis of chiral gold nanoparticles. |
| 1. |  | Li-Zhe Feng, Jing-Jing Wang, Tao Ma, Yi-Chen Yin, Kuang-Hui Song, Zi-Du Li, Man-Man Zhou, Shan Jin, Taotao Zhuang, Fengjia Fan, Manzhou Zhu, Hong-Bin Yao Biomimetic non-classical crystallization drives hierarchical structuring of efficient circularly polarized phosphors In: Nature Communications, vol. 13, no. 3339, 2022, (Chiral emitters with high photoluminescence quantum yield are desirable for use in circularly polarized LEDs. The authors demonstrate the transfer of chirality from nanoscale copper iodide clusters to microscale chiral luminescent polycrystals by non-classical crystallization.). @article{nokey,
title = {Biomimetic non-classical crystallization drives hierarchical structuring of efficient circularly polarized phosphors},
author = {Li-Zhe Feng and Jing-Jing Wang and Tao Ma and Yi-Chen Yin and Kuang-Hui Song and Zi-Du Li and Man-Man Zhou and Shan Jin and Taotao Zhuang and Fengjia Fan and Manzhou Zhu and Hong-Bin Yao},
editor = {Christian Kuttner (Ed.)},
url = {https://www.nature.com/articles/s41467-022-30989-y.pdf},
doi = {10.1038/s41467-022-30989-y},
year = {2022},
date = {2022-06-09},
urldate = {2022-06-09},
journal = {Nature Communications},
volume = {13},
number = {3339},
abstract = {Hierarchically structured chiral luminescent materials hold promise for achieving efficient circularly polarized luminescence. However, a feasible chemical route to fabricate hierarchically structured chiral luminescent polycrystals is still elusive because of their complex structures and complicated formation process. We here report a biomimetic non-classical crystallization (BNCC) strategy for preparing efficient hierarchically structured chiral luminescent polycrystals using well-designed highly luminescent homochiral copper(I)-iodide hybrid clusters as basic units for non-classical crystallization. By monitoring the crystallization process, we unravel the BNCC mechanism, which involves crystal nucleation, nanoparticles aggregation, oriented attachment, and mesoscopic transformation processes. We finally obtain the circularly polarized phosphors with both high luminescent efficiency of 32% and high luminescent dissymmetry factor of 1.5 × 10^−2, achieving the demonstration of a circularly polarized phosphor converted light emitting diode with a polarization degree of 1.84% at room temperature. Our designed BNCC strategy provides a simple, reliable, and large-scale synthetic route for preparing bright circularly polarized phosphors.},
note = {Chiral emitters with high photoluminescence quantum yield are desirable for use in circularly polarized LEDs. The authors demonstrate the transfer of chirality from nanoscale copper iodide clusters to microscale chiral luminescent polycrystals by non-classical crystallization.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hierarchically structured chiral luminescent materials hold promise for achieving efficient circularly polarized luminescence. However, a feasible chemical route to fabricate hierarchically structured chiral luminescent polycrystals is still elusive because of their complex structures and complicated formation process. We here report a biomimetic non-classical crystallization (BNCC) strategy for preparing efficient hierarchically structured chiral luminescent polycrystals using well-designed highly luminescent homochiral copper(I)-iodide hybrid clusters as basic units for non-classical crystallization. By monitoring the crystallization process, we unravel the BNCC mechanism, which involves crystal nucleation, nanoparticles aggregation, oriented attachment, and mesoscopic transformation processes. We finally obtain the circularly polarized phosphors with both high luminescent efficiency of 32% and high luminescent dissymmetry factor of 1.5 × 10^−2, achieving the demonstration of a circularly polarized phosphor converted light emitting diode with a polarization degree of 1.84% at room temperature. Our designed BNCC strategy provides a simple, reliable, and large-scale synthetic route for preparing bright circularly polarized phosphors. |

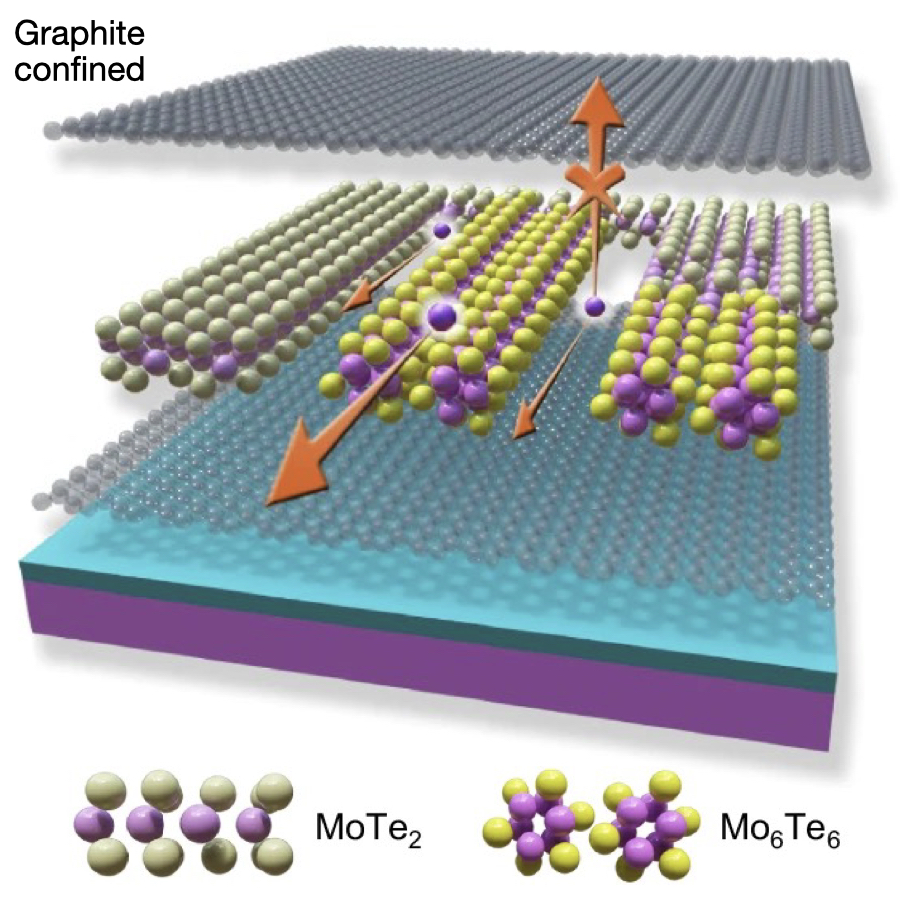
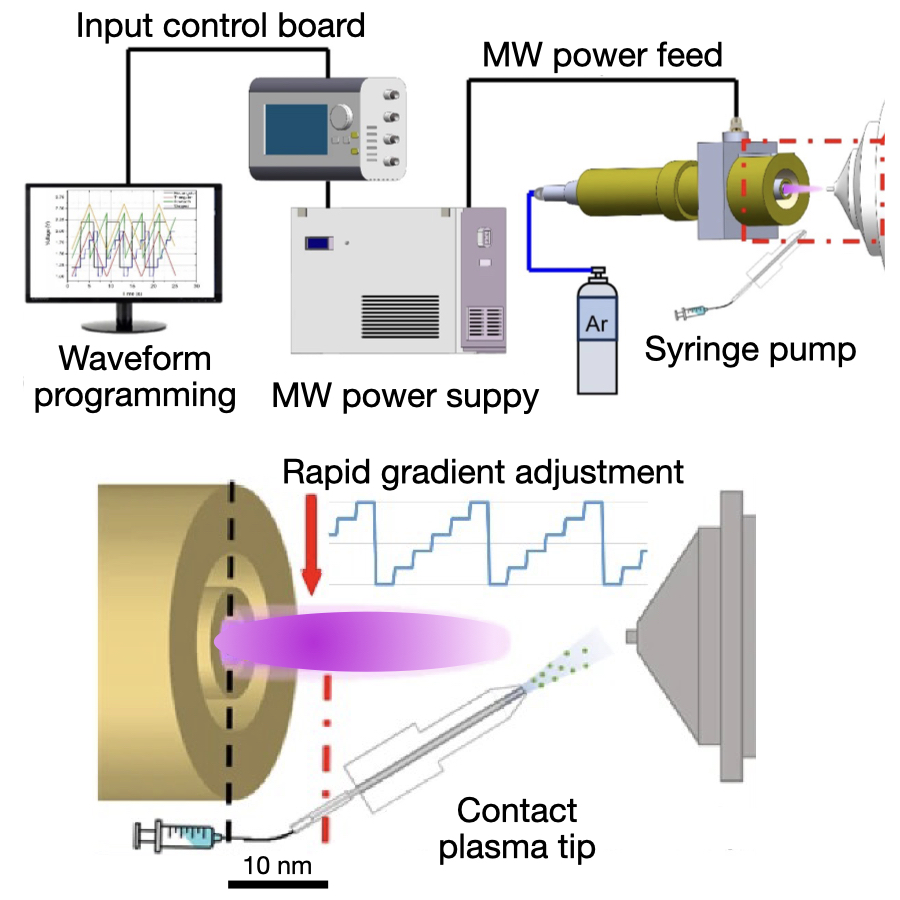
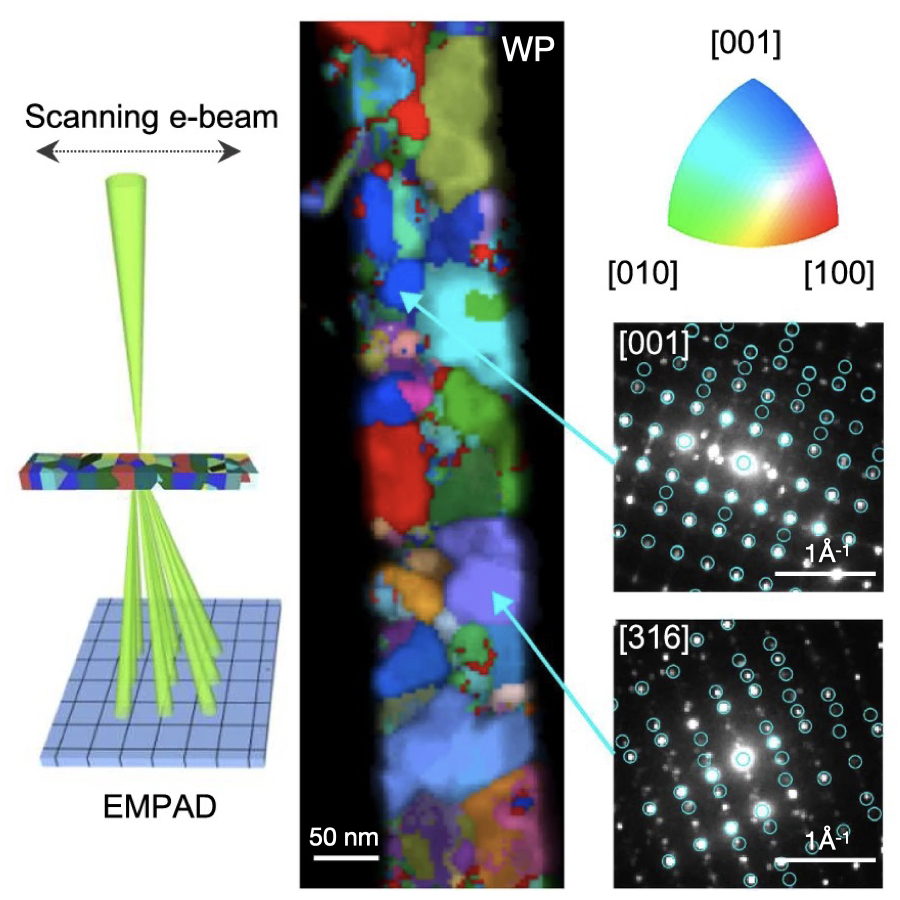
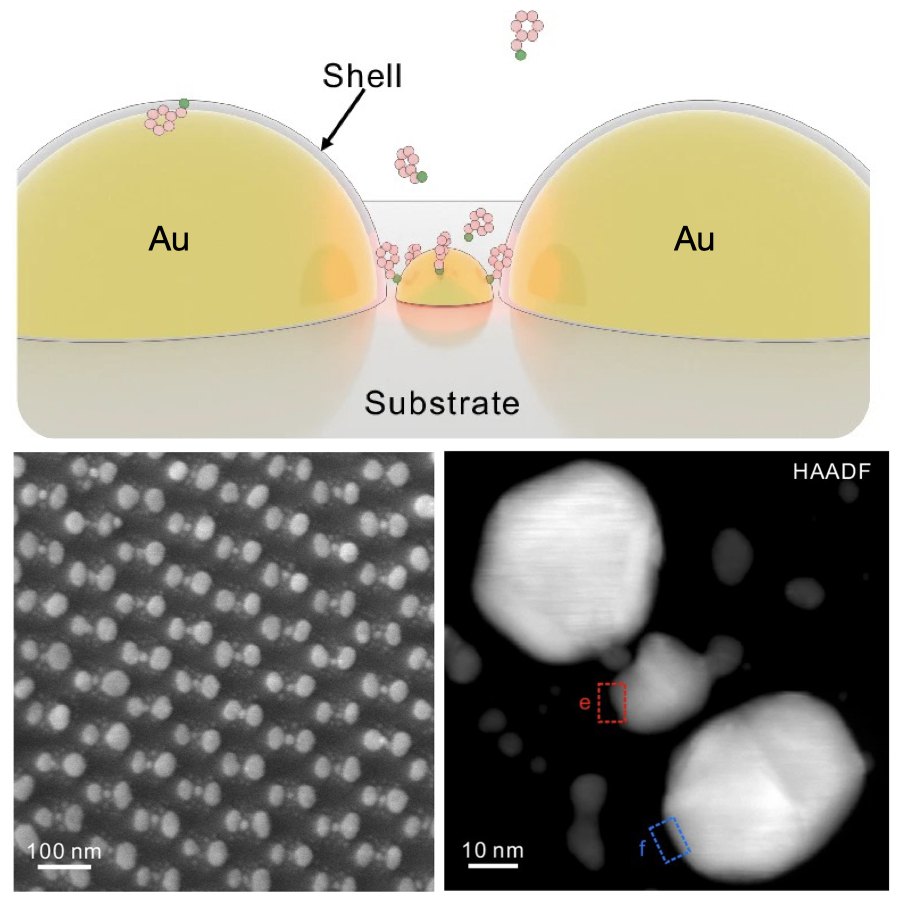
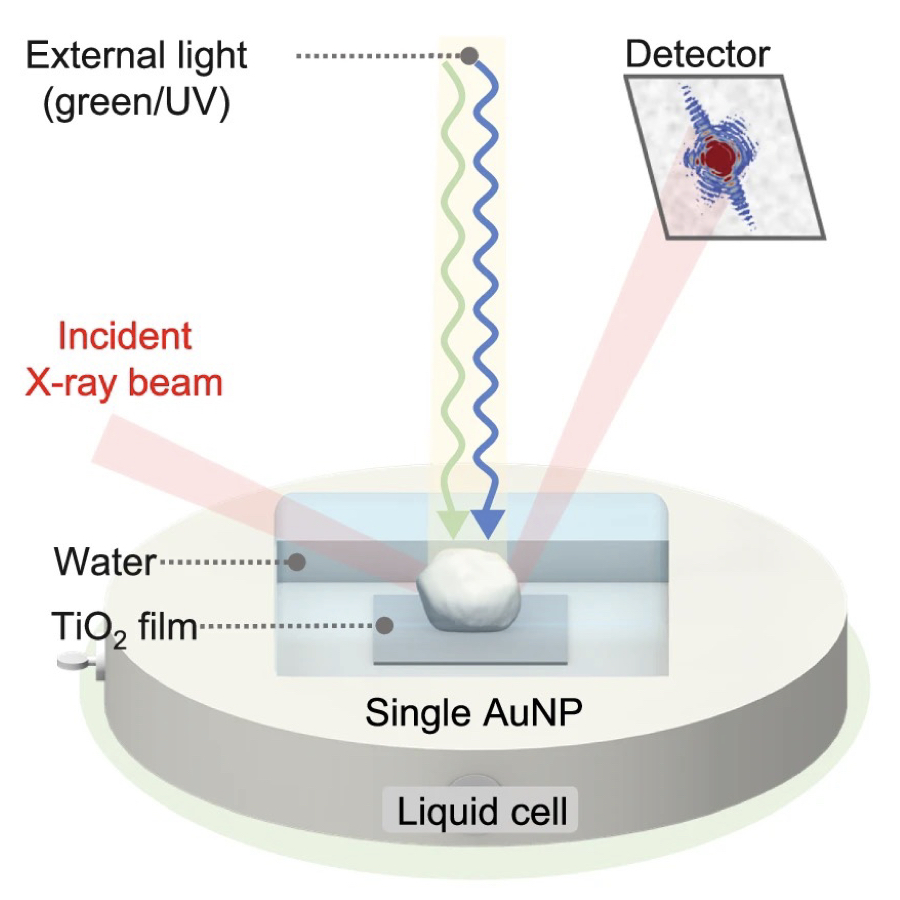

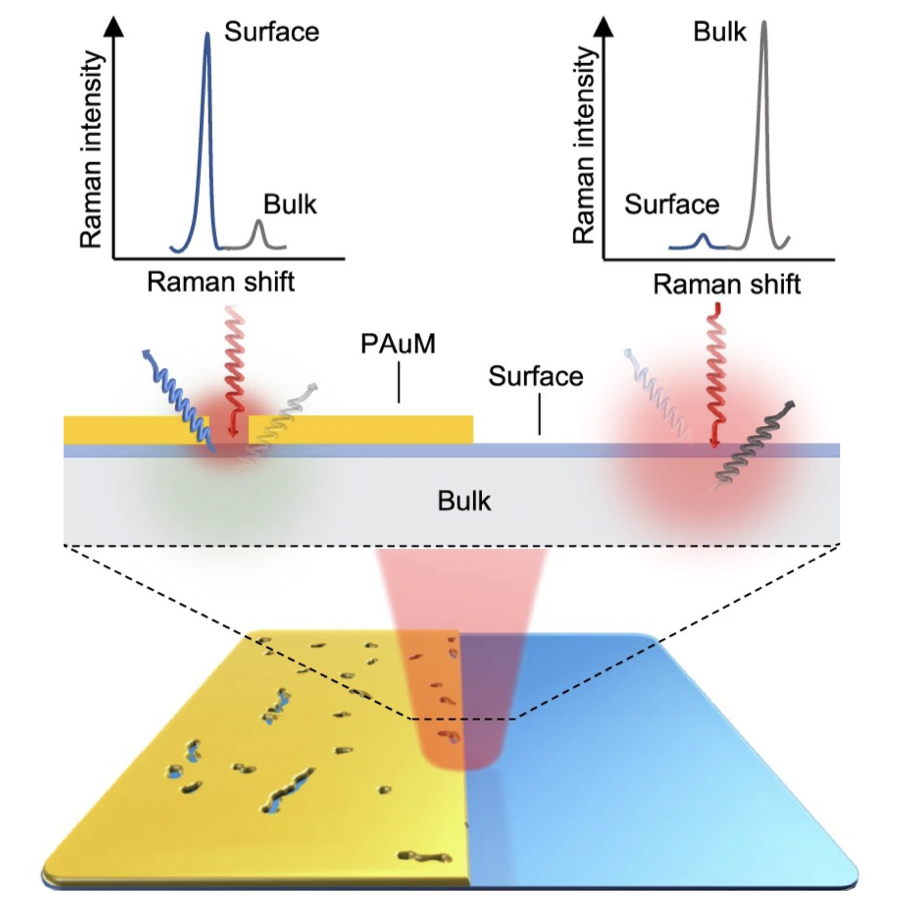
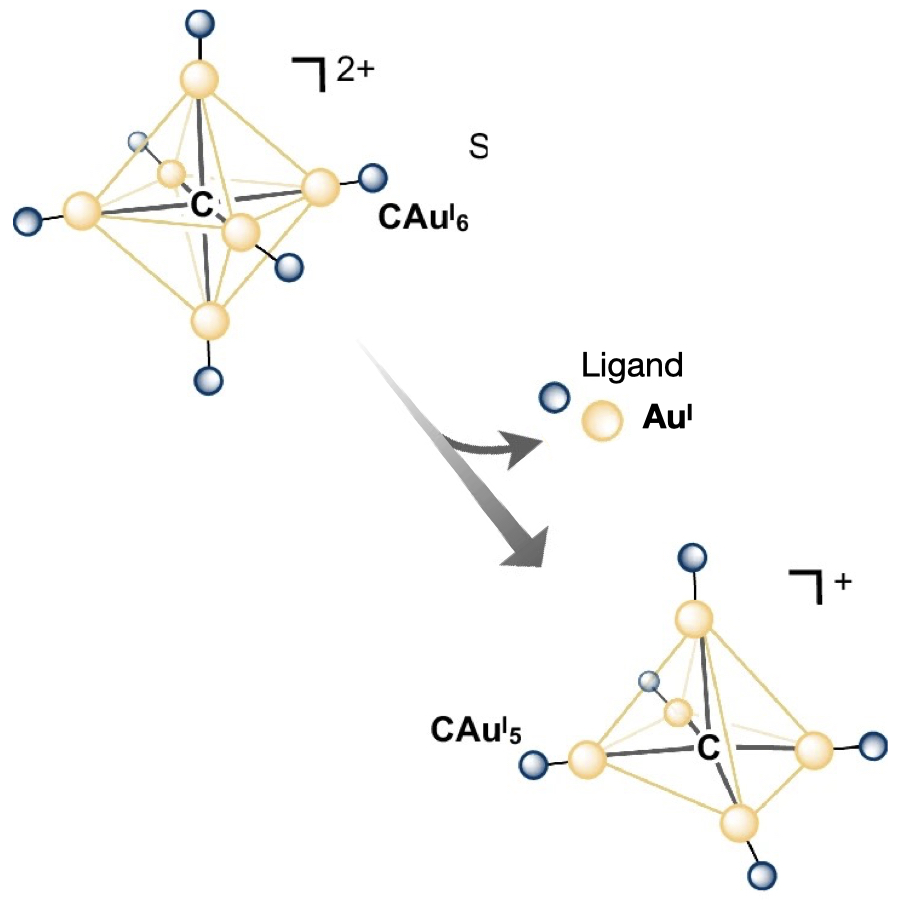

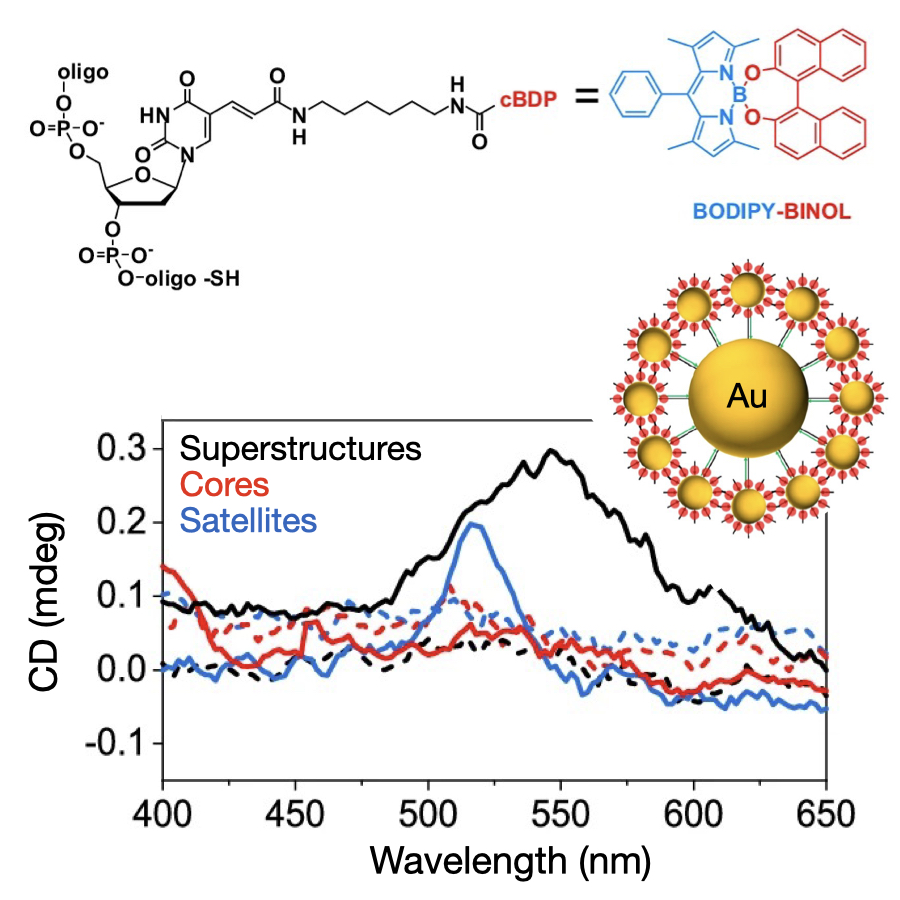

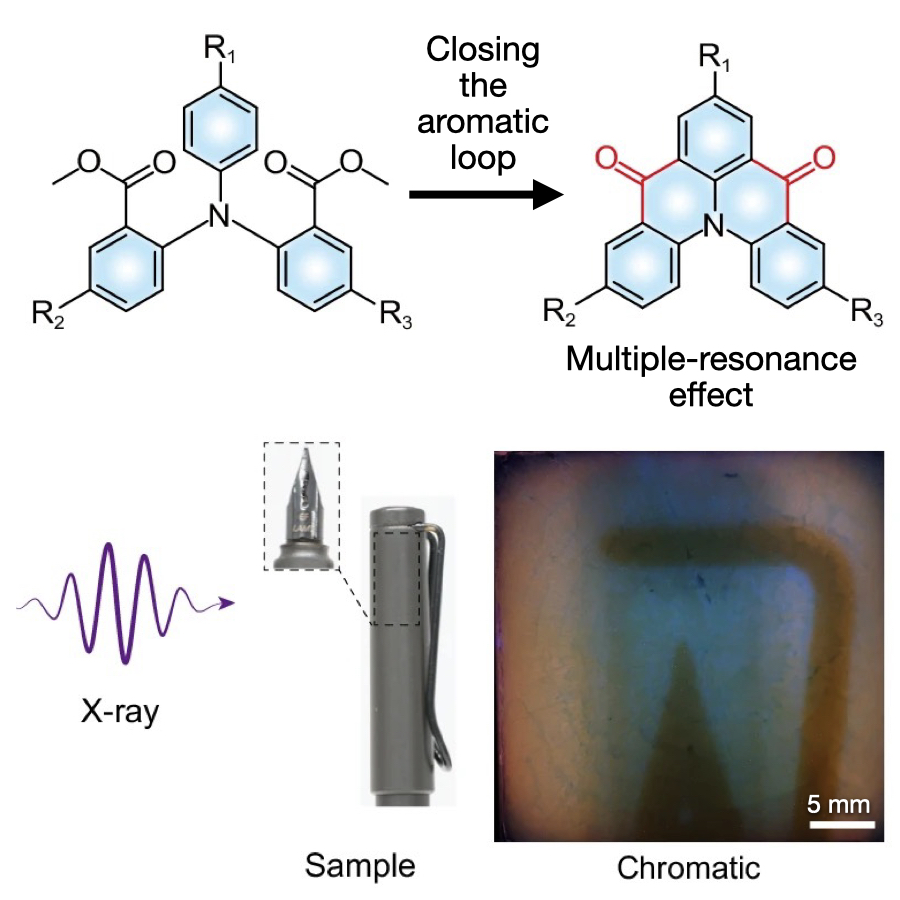



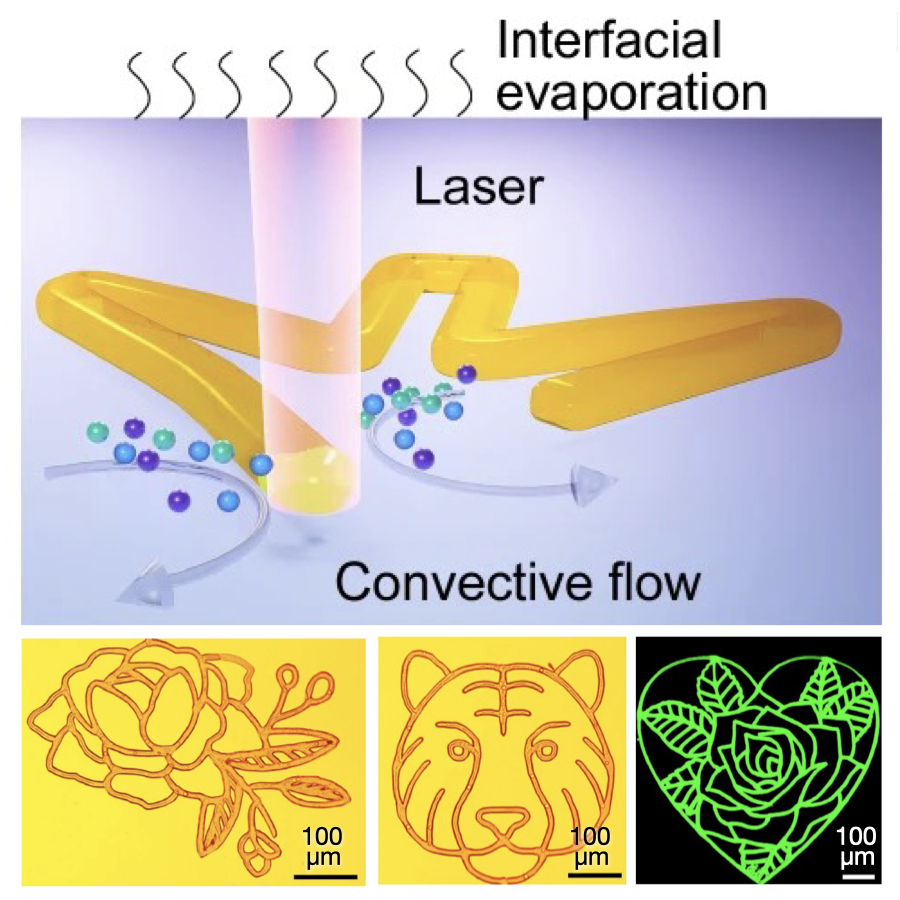





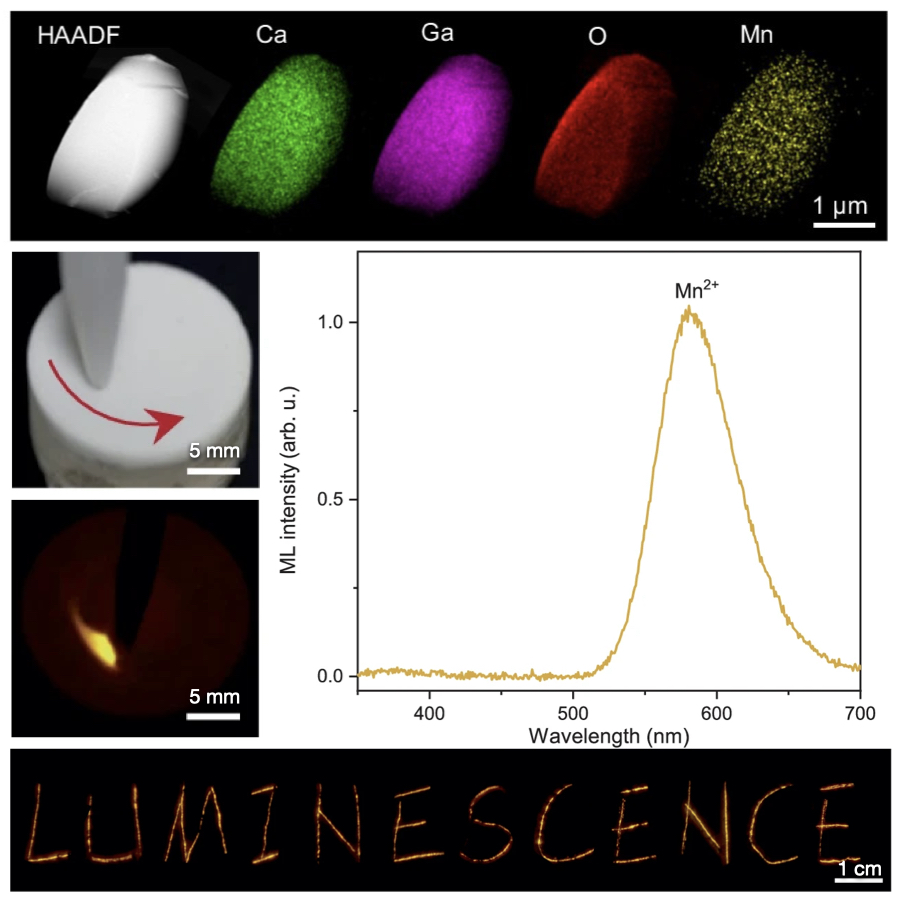


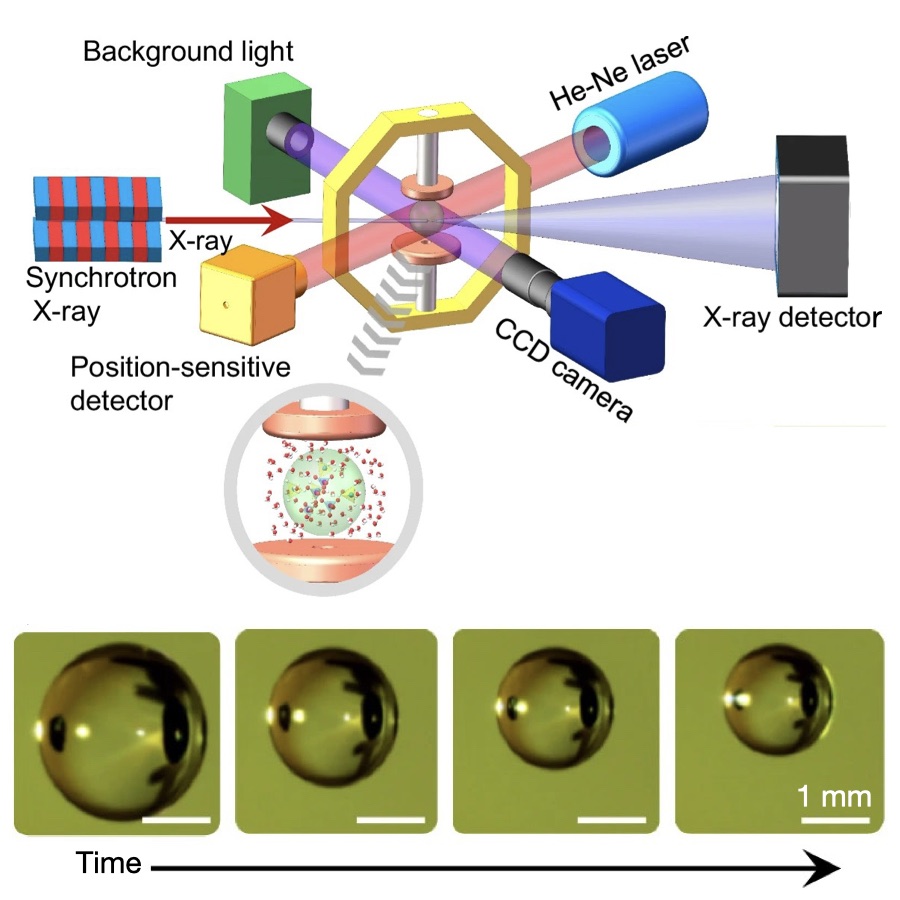












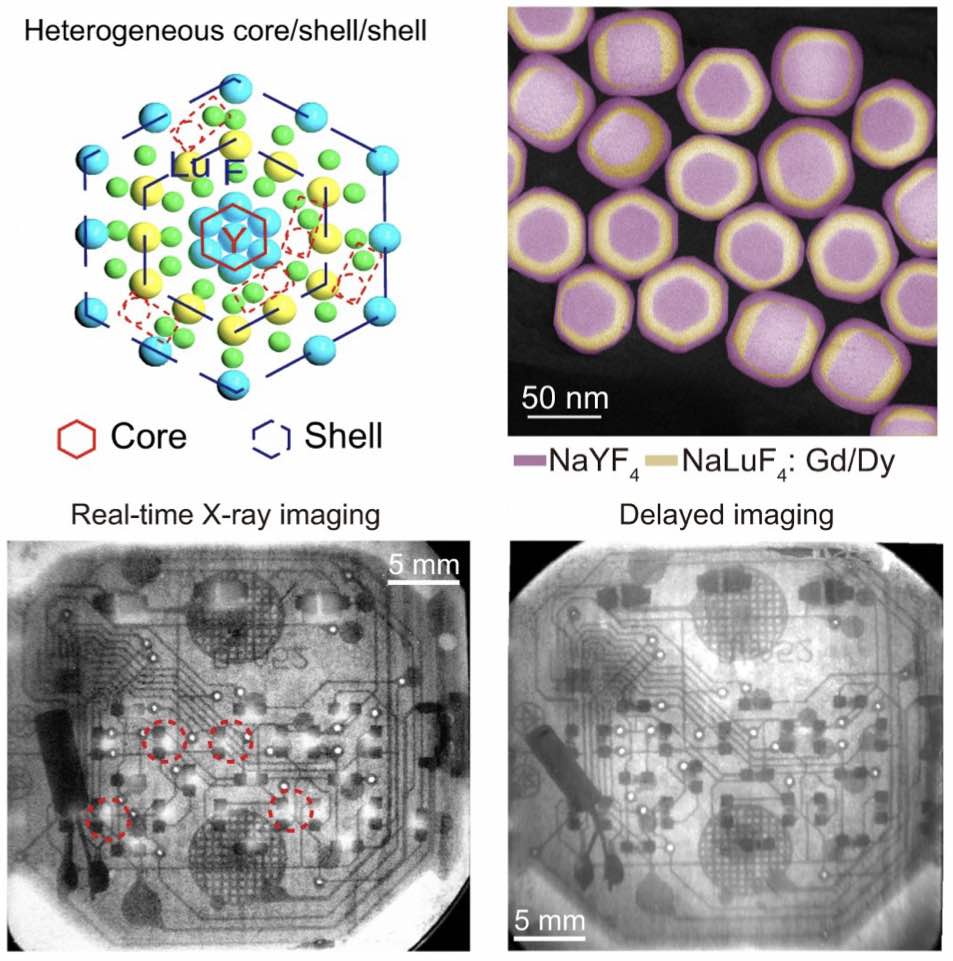
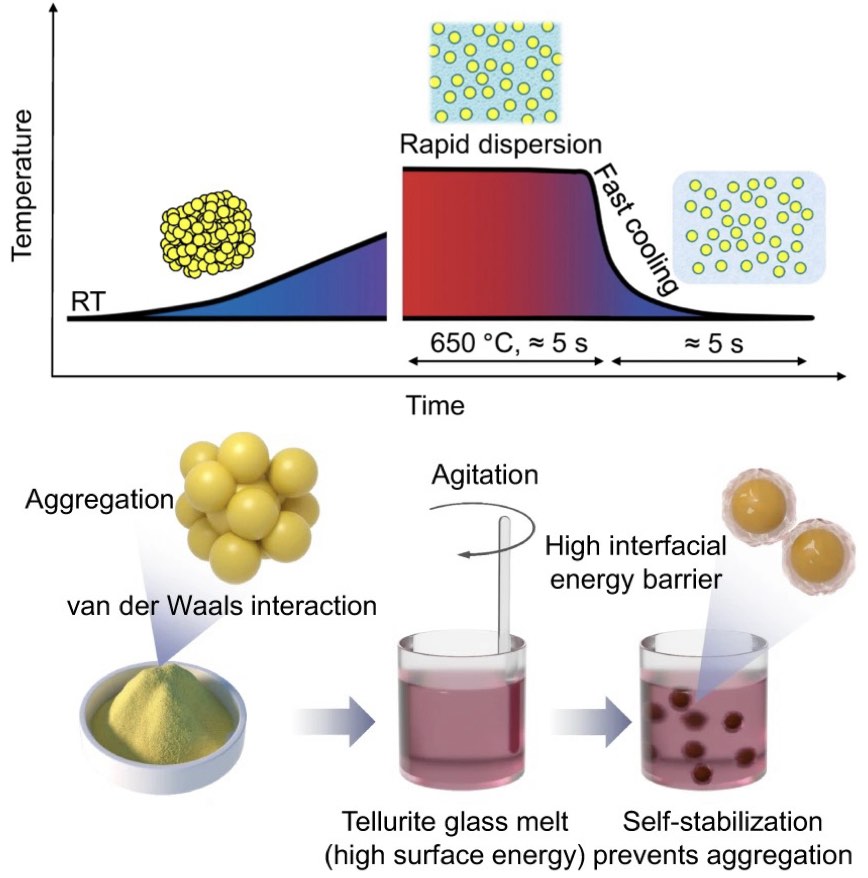


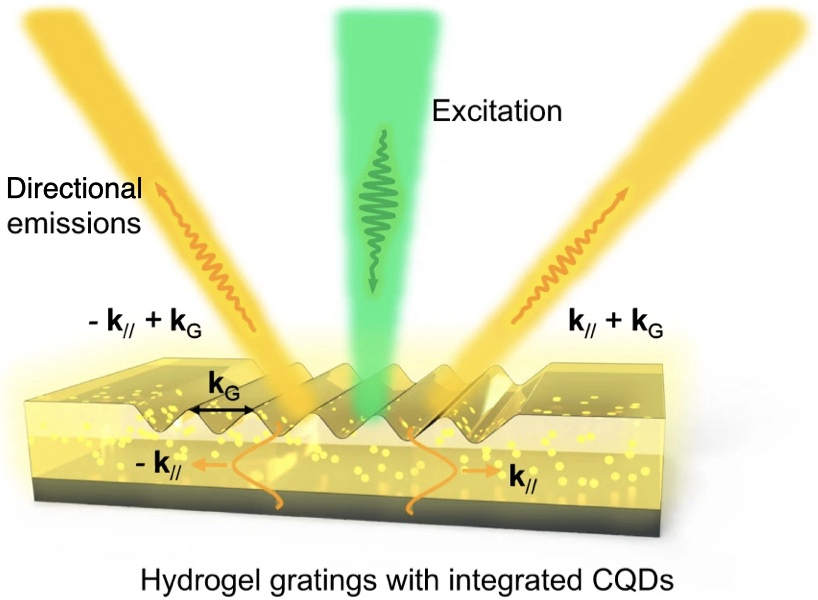
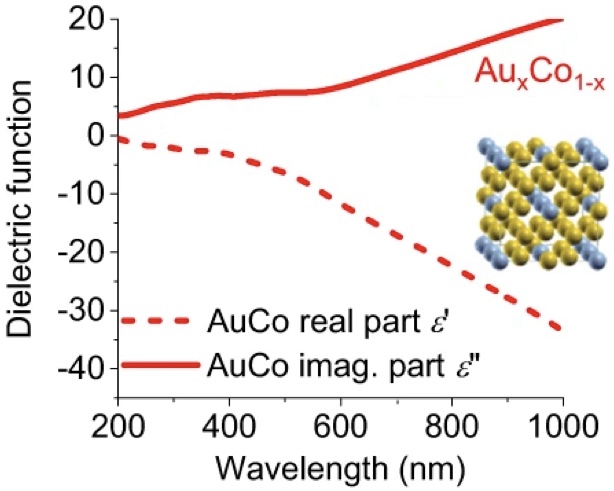
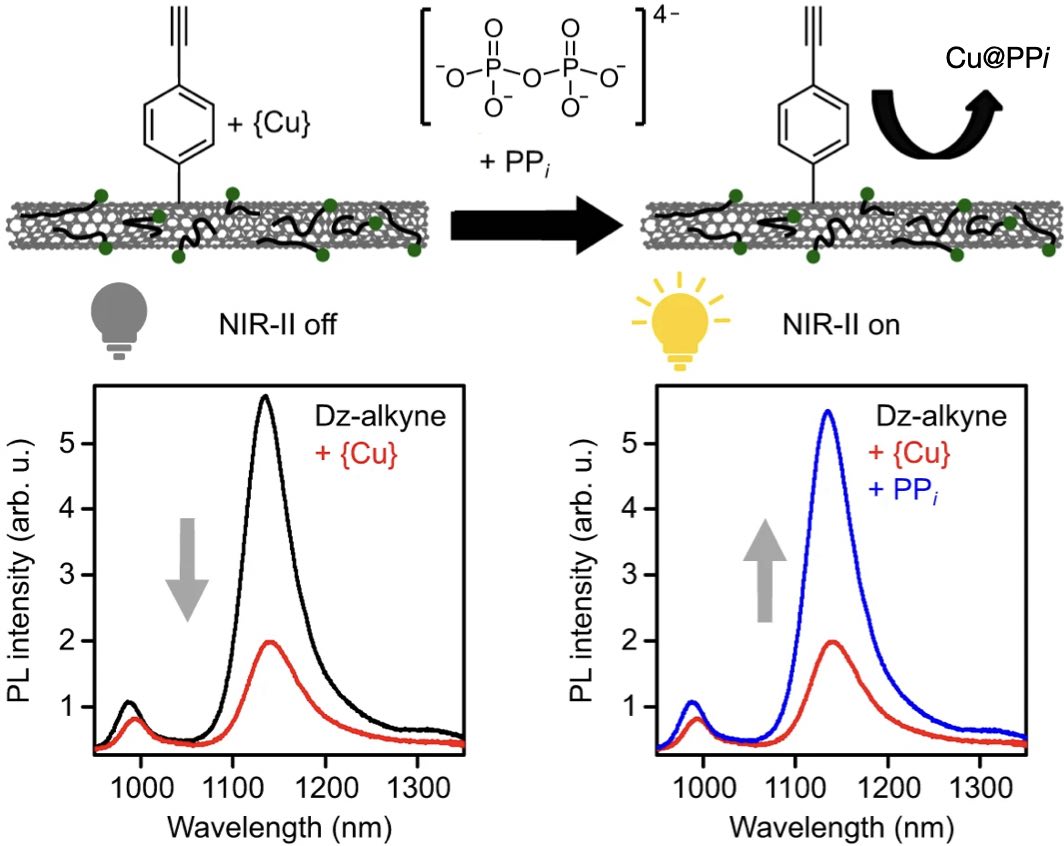
![Synthesis of inter-[60]fullerene conjugates with inherent chirality](https://christiankuttner.de/wp-content/uploads/2024/02/TOC113.jpeg)